This is an easy-to-use bedside calculation that operates as an outcome predictor in a way that is strikingly similar to the measured physiological dead-space fraction. Higher values indicate greater ventilatory inefficiency and a higher dead-space fraction
[32]. It has been shown to be capable of predicting mortality independently of oxygenation, shock status, vasopressor use, and Acute Physiology and Chronic Health Evaluation III (APACHE-III) scores
[33][34][35], with odds ratios comparable to those of Nuckton and colleagues
[33], thereby outperforming empirical estimates, except with respect to direct estimation
[29]. The trajectory of ventilatory ratios during the early stages of ARDS has also been associated with survival and may prove valuable when tracking a patient’s clinical course
[29][36].
Interestingly, in a recent secondary analysis of the large (
n = 927) PRoVENT-COVID study cohort, none of the previously discussed formulae for dead-space estimation, nor the ventilatory ratio, were found to be independent predictors of mortality in an adjusted base risk model. Only direct estimation—when measured early at the start of ventilation, but not on the following day—retained additional prognostic value. The researchers also argued that in patients with COVID-19, dead-space might be regarded more as a marker of ARDS severity rather than as an independent prognostication factor. Despite its apparent simplicity, the P
ETCO
2/PaCO
2 ratio was the only index in this cohort to be independently associated with outcomes both at the start of ventilation and on day one
[28] and has been recently validated as a predictor of mortality in a large retrospective cohort
[37].
3. Hypoxic Pulmonary Vasoconstriction and V/Q Mismatch as Mechanisms of VILI
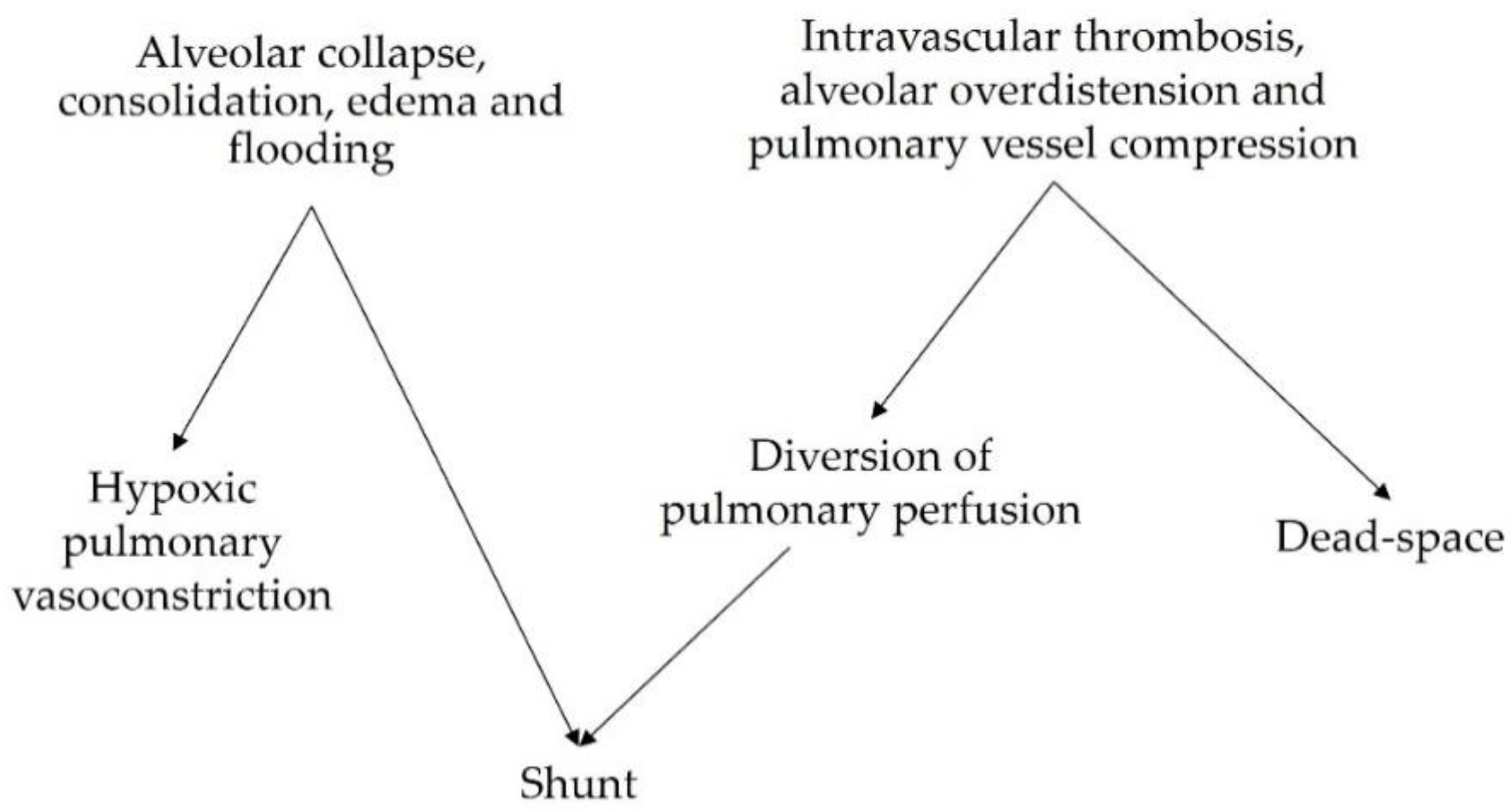
Beyond causing impairments in gas exchange, V/Q mismatch and the physiologic responses to mismatch have been implicated in the development and progression of lung injury in patients with ARDS. A summary of the corresponding explanatory mechanisms is presented in Table 1.
Table 1. V/Q mismatch as a mechanism of lung injury.
3.1. VILI Related to Perfused Non-Ventilated Lung Units
The perfusion of low
V/
Q and shunted lung zones in healthy lungs is counterbalanced by hypoxic pulmonary vasoconstriction (HPV), a physiological response to alveolar hypoxia and/or to decreased mixed venous oxygen content that diverts pulmonary blood flow from poorly ventilated to more normally ventilated lung zones
[54], thereby improving
V/
Q matching and gas exchange. HPV has been extensively investigated in experimental studies. In ARDS models induced by E. Coli endotoxin and oleic acid infusion, HPV appeared to be inhibited
[39][40][55][56], contributing to increased shunt and worsening oxygenation. In other models, induced mainly by oleic acid infusion, HPV was preserved
[41]. In ARDS patients, the effectiveness of HPV might vary depending on etiology, hemodynamic status, the administration of medications, and preexisting pulmonary conditions. However, both experimental
[42] and clinical
[43] observations of worsening acute pulmonary hypertension induced by hypoxemia and acidosis, and of the hypoxemic effects of intravenous vasodilators, suggest that HPV is preserved in most ARDS patients.
The observation that intrapulmonary shunting and physiological HPV redistribute pulmonary perfusion has shed light on the potentially deleterious effects of hypo-perfused lung zones in the genesis and/or progression of lung injury in ARDS. Studies of healthy lungs demonstrated that HPV optimizes gas exchange but may also limit the supply of oxygen and nutrients to shunted lung zones
[44]. In the presence of atelectasis, the size of the aerated lung is decreased, thus increasing the risk of overdistension and barotrauma of the ventilated areas and, consequently, promoting inflammation
[45]. SPECT analysis of pigs undergoing one-lung ventilation (OLV) for 90 min followed by two-lung ventilation (TLV) showed hyperinflation and hyperperfusion of the ventilated lung, which caused diffuse damage to the alveolar compartment
[46]. However, most studies of OLV models in the literature were performed for only a few hours and focused on the development of lung injury after TLV was restored (ischemia-reperfusion injury). Recently, the impact of OLV with a higher and lower V
T for 24 h without restoration of TLV was studied in pigs by group
[47] (
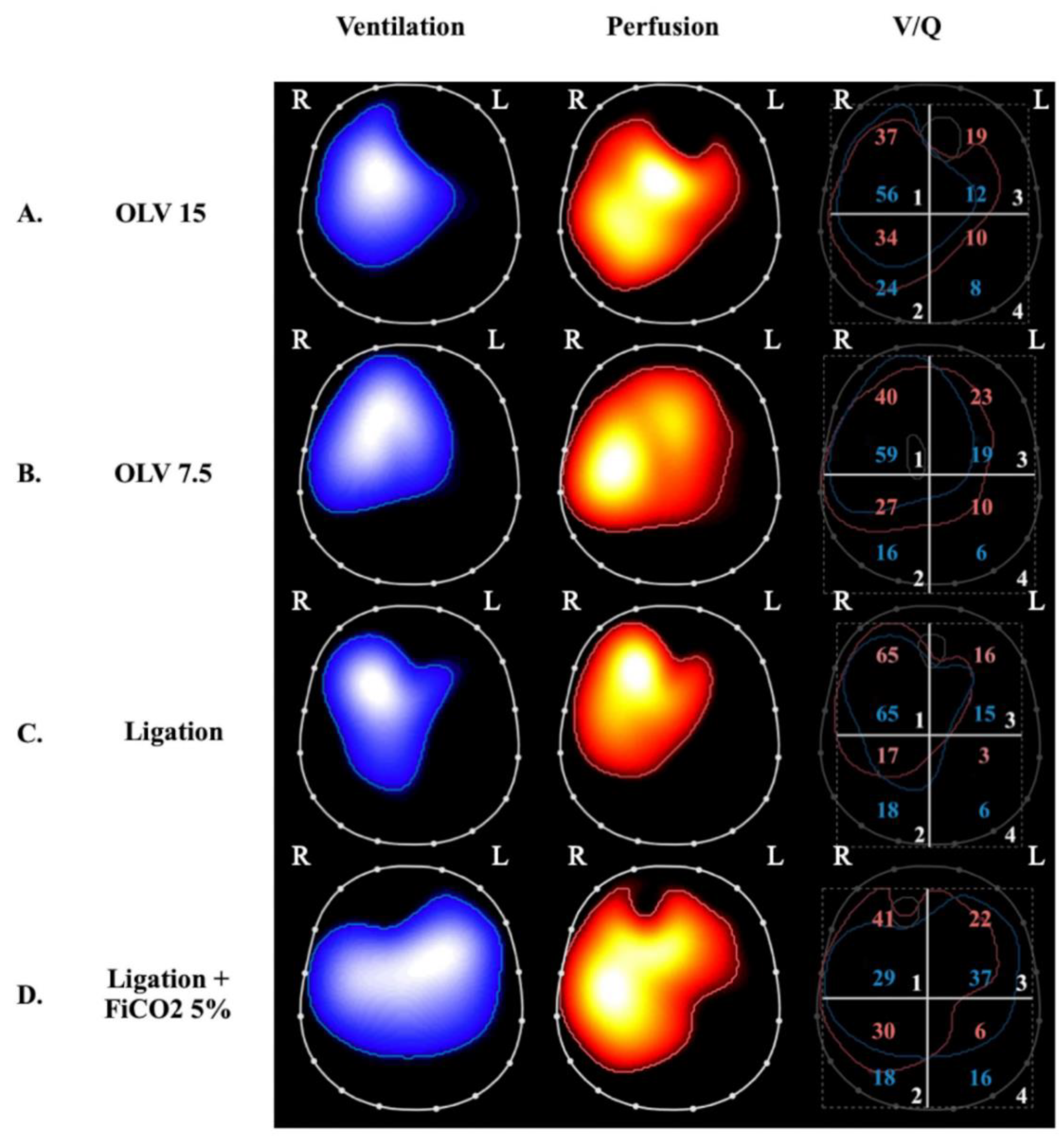
Figure 2).
Figure 2. Ventilation, perfusion, and ventilation-perfusion matching via electrical impedance tomography in 4 experimental swine study groups. Images on the left display regional ventilation, middle images depict regional perfusion and were obtained by administering a hypertonic saline bolus under apneic conditions (see text), and images on the right depict ventilation-perfusion matching, which is expressed as the superposition of the ventilation and perfusion maps. The percentages of ventilation and perfusion to each of the four quadrants are annotated as blue and red numbers, respectively, on the right-side panels. The letters R and L indicate the right and left lung, respectively. (Panels
A and
B) were obtained during one-lung ventilation (OLV) with exclusion of the left lung and a tidal volume of 15 mL/kg (panel
A) and 7.5 mL/kg (panel
B)
[47]. At both tidal volumes, there is no ventilation of the left lung and perfusion appears to be redistributed to the ventilated lung. OLV at higher tidal volume (panel
A) caused bilateral lung injury (lung histological score 5 ± 2 in the right lung and 10 ± 2 in the left lung); this was compared to two-lung-ventilated controls (lung histological score 3 ± 1 in right lung and 3 ± 1 in left lung). Interestingly, lowering tidal volume to 7.5 mL/kg (panel B) attenuated inflammation and lung injury (lung histological score 3 ± 1 in the right lung and 7 ± 1 in the left lung) despite an absence of change in the overall distributions of ventilation and perfusion (ANOVA
p ≤ 0.01 for the right lung and
p ≤ 0.001 for the left lung). (Panels
C and
D) were obtained from a study of selective left pulmonary artery ligation
[48]. (Panel
C) represents ligation alone whereas (panel
D) represents ligation + 5% inhaled CO
2. The two groups differ significantly both for ventilation and perfusion distributions: In the ligation group, perfusion is only present in the right lung, ventilation is also diverted to the right lung, and total lung histological score was 11 ± 3. In the ligation + inhaled CO
2 group, there is a more homogeneous distribution of ventilation and perfusion in both lungs and total lung histological score decreased to 4 ± 2 (ANOVA
p ≤ 0.0001). The occurrence of perfusion to the ligated lung with inhaled CO
2 is thought to transpire due to increased flow through bronchial circulation.
OLV caused bilateral lung injury and, interestingly, lowering VT prevented injury to the ventilated lung, but this protection was only partial in the non-ventilated lung. EIT analyses demonstrated that lung stress (classical VILI mechanism) was the main mechanism of injury in the ventilated lungs but collapse and hypoperfusion were implicated in the non-ventilated lungs. Notably, inflammation measured in the non-ventilated lung was dampened by reducing VT to the contralateral ventilated lung, indicating the possible role of inflammation-based crosstalk between the lungs.
3.2. VILI Related to Ventilated Non-Perfused Lung Units
The other extreme of
V/
Q mismatch is represented by areas with high and infinite
V/
Q (dead-space), which have been studied in experimental settings mostly by intravascular occlusion or the surgical ligation of the pulmonary arteries. Alveolar hypocapnia in ventilated non-perfused alveoli seems to be responsible for local lung injury, which is, in part, mediated by alterations of the surfactant system
[49] that lead to alveolar damage due to instability
[50][51][52][53][57][58] and apoptosis
[59]. Non-perfused ventilated areas can develop hemorrhagic infarction, as shown in a model of cardiopulmonary bypass and preserved ventilation
[60]. Healthy swine lungs undergoing regional pulmonary vascular occlusion were studied to assess the local diversion of V
T from nonperfused to perfused areas via computed tomography. This diversion of ventilation appeared to be a compensatory mechanism that effectively limits
V/
Q mismatch
[61] but also an indirect mechanism of lung injury due to overdistention and injury to perfused ventilated regions (
Table 1 and
Figure 2, panel C). As with HPV for perfusion, hypocapnic bronchoconstriction can divert ventilation, improve
V/
Q matching, and optimize gas exchange by redistributing V
T to better-perfused lung areas
[62][63]; on the other hand, it may contribute to the development of injury in these areas due to concomitant hyperventilation and hyperperfusion
[64]. In a model of pulmonary hypertension induced by E. Coli endotoxin in sheep, it was shown that there is no threshold for edema formation when capillary permeability was increased; any increase in pulmonary blood flow or pressure increased edema
[65]. This finding supports the hypothesis that an increase in regional lung perfusion in conditions of local injury (for example, due to increased lung stress from hyperventilation) can result in further injury due to edema formation. Furthermore, mild bilateral lung injury that develops in dogs after unilateral pulmonary artery occlusion is characterized by endothelial abnormalities and perivascular edema
[66]. Broccard et al. observed that the dependent distribution of VILI in supine large animals ventilated with high V
T might be explained by regional differences in blood flow and vascular pressure, suggesting that differences among ventilatory patterns may be due, at least partially, to differences in hemodynamics
[67].
Since the 1960s, the role of alveolar hypocapnia in the development of lung injury in non-perfused ventilated lung units has been demonstrated by studying the impact of inhaled CO
2 in unilateral pulmonary artery ligation (UPAL) models. Edmunds et al. first found a significant reduction in atelectasis in the ligated lung and a local increase in ventilation induced by inhaled CO
2, which led them to hypothesize that there was a direct effect of CO
2 or [H+] on bronchiolar alveolar cells and surfactant
[68]. Kolobow et al. observed a reduction in hemorrhagic infarction and decreased alveolar and capillary injury in spontaneously breathing lambs during total cardiopulmonary bypass coupled with inhaled CO
2. The effect of high PCO
2 inhalation was also studied in preterm lambs; the result was an increase in lung gas volume and a reduction in histological damage and inflammation
[69]. Recently, the group described a significant reduction in bilateral lung injury due to left-sided UPAL by administering 5% inhaled CO
2 during controlled mechanical ventilation
[48] (
Figure 2). The findings confirmed the protection of the ligated lung but also highlighted the protective role of inhaled CO
2 in the non-ligated lung, which was less overdistended due to a more homogenous distribution of ventilation as assessed by EIT. The group also investigated the question of whether the protective effects of inhaled CO
2 in the setting of bilateral lung injury caused by UPAL were due to increased PaCO
2 or to the local effects of CO
2 inhalation. Researchers demonstrated that inhaled CO
2 allows for more effective bilateral lung protection compared to plasmatic hypercapnia obtained through other methods
[70].
In summary, in the presence of an elevated dead-space fraction, ventilated non-perfused units can be damaged by the inhibition of surfactant production and function, the induction of apoptosis, local ischemia, and inflammatory crosstalk from the residual hyperventilated and hyperperfused lung.
 +1 credit
+1 credit