Acute respiratory distress syndrome (ARDS) is a heterogenous condition that is characterized by the development of inflammatory pulmonary edema and life-threatening hypoxemia, and it accounts for nearly 25% of patients who require mechanical ventilation. Acute respiratory distress syndrome (ARDS) remains an important clinical challenge with a mortality rate of 35–45%. It is being increasingly demonstrated that the improvement of outcomes requires a tailored, individualized approach to therapy, guided by a detailed understanding of each patient’s pathophysiology. In patients with ARDS, disturbances in the physiological matching of alveolar ventilation (V) and pulmonary perfusion (Q) (
V
/
Q
mismatch) are a hallmark derangement. The perfusion of collapsed or consolidated lung units gives rise to intrapulmonary shunting and arterial hypoxemia, whereas the ventilation of non-perfused lung zones increases physiological dead-space, which potentially necessitates increased ventilation to avoid hypercapnia.
- electrical impedance tomography
- acute respiratory distress syndrome
- ventilation-induced lung injury
1. Genesis of Ventilation-Perfusion Mismatch in the Acute Respiratory Distress Syndrome
1.1. Non-Ventilated Perfused Units (Shunt)
1.2. Ventilated Non-Perfused Units (Dead-Space)
2. V/Q Mismatch as a Marker of Severity in ARDS Patients
2.1. Intrapulmonary Shunt
2.2. Dead-Space
2.3. Assessment of V/Q Mismatch by EIT
The sum of the percentages of non-perfused ventilated and perfused non-ventilated lung units (unmatched units) obtained by superimposing EIT-derived ventilation and perfusion maps has recently been shown to be capable of independently predicting mortality in ARDS patients. A value of 27% unmatched units predicted mortality with a positive predictive value of 67% and a negative predictive value of 91%. The percentage of only perfused units (i.e., an estimate of a shunt) was significantly inversely correlated with the PaO2/FiO2 ratio and the dorsal fraction of ventilation [38][61]. These findings represent an additional means to assess ARDS severity at the bedside and lay the foundation for more advanced analyses. EIT imaging offers the potential to differentiate between dead-space and shunt fractions, potentially bridging the gap between bedside measures and respiratory pathophysiology.3. Hypoxic Pulmonary Vasoconstriction and V/Q Mismatch as Mechanisms of VILI
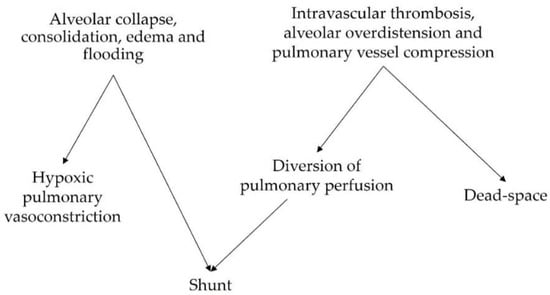
Beyond causing impairments in gas exchange, V/Q mismatch and the physiologic responses to mismatch have been implicated in the development and progression of lung injury in patients with ARDS. A summary of the corresponding explanatory mechanisms is presented in Table 1.| Mechanisms of Injury | Reference | ||
|---|---|---|---|
| Shunt (perfused non-ventilated lung units) | Redistribution of perfusion due to hypoxic pulmonary vasoconstriction: hypo-perfused lung zones with locally decreased oxygen and nutrient delivery and lung ischemia. | [39] | [75] |
| Decreased size of aerated lung with increased risk of overdistension and barotrauma in the ventilated lung. | [40][41][42] | [76,77,78] | |
| Dead-space (ventilated non-perfused lung units) | Local alveolar hypocapnia: altered surfactant system, alveolar instability, apoptosis, and hemorrhagic infarction. | [43][44][45][46][47][48][49][50][51] | [79,80,81,82,83,84,85,86,87] |
| Local bronchoconstriction with diversion of ventilation to perfused lung zones resulting in hyperventilation and hyperperfusion in diverted zones. | [52][53] | [88,89] |
3.1. VILI Related to Perfused Non-Ventilated Lung Units
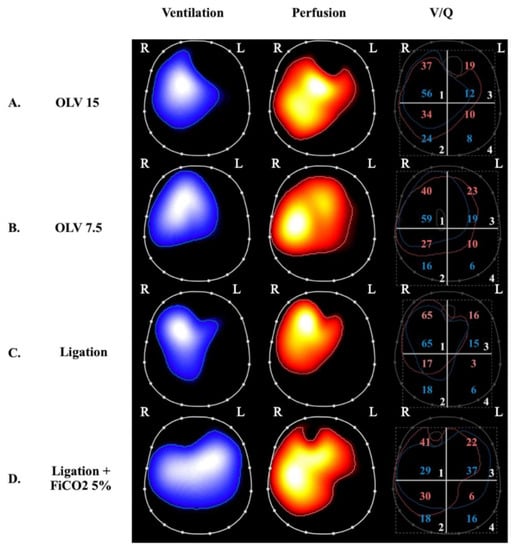
The perfusion of low V/Q and shunted lung zones in healthy lungs is counterbalanced by hypoxic pulmonary vasoconstriction (HPV), a physiological response to alveolar hypoxia and/or to decreased mixed venous oxygen content that diverts pulmonary blood flow from poorly ventilated to more normally ventilated lung zones [54][90], thereby improving V/Q matching and gas exchange. HPV has been extensively investigated in experimental studies. In ARDS models induced by E. Coli endotoxin and oleic acid infusion, HPV appeared to be inhibited [39][40][55][56][75,76,91,92], contributing to increased shunt and worsening oxygenation. In other models, induced mainly by oleic acid infusion, HPV was preserved [41][77]. In ARDS patients, the effectiveness of HPV might vary depending on etiology, hemodynamic status, the administration of medications, and preexisting pulmonary conditions. However, both experimental [42][78] and clinical [43][79] observations of worsening acute pulmonary hypertension induced by hypoxemia and acidosis, and of the hypoxemic effects of intravenous vasodilators, suggest that HPV is preserved in most ARDS patients. The observation that intrapulmonary shunting and physiological HPV redistribute pulmonary perfusion has shed light on the potentially deleterious effects of hypo-perfused lung zones in the genesis and/or progression of lung injury in ARDS. Studies of healthy lungs demonstrated that HPV optimizes gas exchange but may also limit the supply of oxygen and nutrients to shunted lung zones [44][80]. In the presence of atelectasis, the size of the aerated lung is decreased, thus increasing the risk of overdistension and barotrauma of the ventilated areas and, consequently, promoting inflammation [45][81]. SPECT analysis of pigs undergoing one-lung ventilation (OLV) for 90 min followed by two-lung ventilation (TLV) showed hyperinflation and hyperperfusion of the ventilated lung, which caused diffuse damage to the alveolar compartment [46][82]. However, most studies of OLV models in the literature were performed for only a few hours and focused on the development of lung injury after TLV was restored (ischemia-reperfusion injury). Recently, the impact of OLV with a higher and lower VT for 24 h without restoration of TLV was studied in pigs by our group [47][83] (Figure 2).