 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sirius Huang | -- | 1346 | 2022-11-08 01:38:10 |
Video Upload Options
Biological crosstalk refers to instances in which one or more components of one signal transduction pathway affects another. This can be achieved through a number of ways with the most common form being crosstalk between proteins of signaling cascades. In these signal transduction pathways, there are often shared components that can interact with either pathway. A more complex instance of crosstalk can be observed with transmembrane crosstalk between the extracellular matrix (ECM) and the cytoskeleton.
1. Crosstalk Between Signalling Pathways
One example of crosstalk between proteins in a signalling pathway can be seen with cyclic adenosine monophosphate's (cAMP) role in regulating cell proliferation by interacting with the mitogen-activated protein (MAP) kinase pathway. cAMP is a compound synthesized in cells by adenylate cyclase in response to a variety of extracellular signals.[1] cAMP primarily acts as an intracellular second messenger whose major intracellular receptor is the cAMP-dependent protein kinase (PKA) that acts through the phosphorylation of target proteins.[2] The signal transduction pathway begins with ligand-receptor interactions extracellularly. This signal is then transduced through the membrane, stimulating adenylyl cyclase on the inner membrane surface to catalyze the conversion of ATP to cAMP.[3][4]
ERK, a participating protein in the MAPK signaling pathway, can be activated or inhibited by cAMP.[5] cAMP can inhibit ERKs in a variety of ways, most of which involve the cAMP-dependent protein kinase (PKA) and the inhibition of Ras-dependent signals to Raf-1.[6] However, cAMP can also stimulate cell proliferation by stimulating ERKs. This occurs through the induction of specific genes via phosphorylation of the transcription factor CREB by PKA.[5] Though ERKs do not appear to be a requirement for this phosphorylation of CREB, the MAPK pathway does play into crosstalk again, as ERKs are required to phosphorylate proteins downstream of CREB.[5] Other known examples of the requirement of ERKs for cAMP-induced transcriptional effects include induction of the prolactin gene in pituitary cells, and of the dopamine beta-hydroxylate gene in pheochromocytomal cells (PC12).[6] A number of diverse mechanisms exist by which cAMP can influence ERK signaling. Most mechanisms involving cAMP inhibition of ERKs uncouple Raf-1 from Ras activation through direct interaction of PKA with Raf-1 or indirectly through PKA interaction with the GTPase Rap1 [6] (see figure 1). PKA may also negatively regulate ERKs by the activation of PTPases. Mechanisms for the activation of ERKs by cAMP are even more varied, usually including Rap1 or Ras, and even cAMP directly.[6]
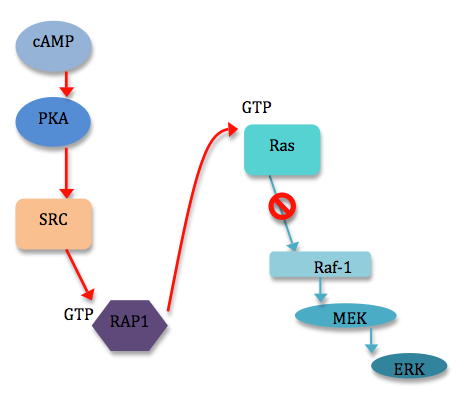
2. Transmembrane Crosstalk
Crosstalk can even be observed across membranes. Membrane interactions with the extracellular matrix (ECM) and with neighboring cells can trigger a variety of responses within the cell. However, the topography and mechanical properties of the ECM also come to play an important role in powerful, complex crosstalk with the cells growing on or inside the matrix.[7] For example, integrin-mediated cytoskeleton assembly and even cell motility are affected by the physical state of the ECM.[7] Binding of the α5β1 integrin to its ligand (fibronectin) activates the formation of fibrillar adhesions and actin filaments.[5] Yet, if the ECM is immobilized, matrix reorganization of this kind and formation of fibrillar adhesions is inhibited.[7] In turn, binding of the same integrin (α5β1) to an immobilized fibronectin ligand is seen to form highly phosphorylated focal contacts/focal adhesion (cells involved in matrix adhesion) within the membrane and reduces cell migration rates[7] In another example of crosstalk, this change in the composition of focal contacts in the cytoskeleton can be inhibited by members of yet another pathway: inhibitors of myosin light-chain kinases or Rho kinases, H-7 or ML-7, which reduce cell contractility and consequently motility.[7] (see figure 2).
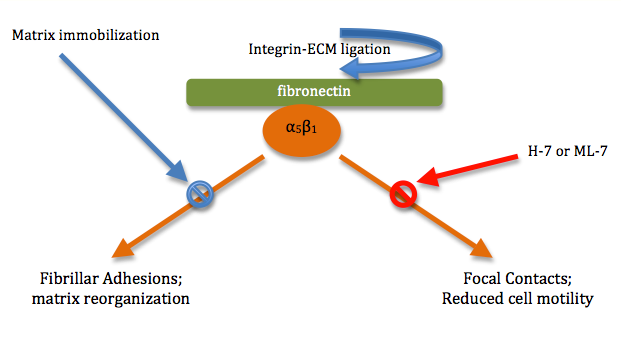
3. Crosstalk in Lymphocyte Activation
A more complex, specific example of crosstalk between two major signaling pathways can be observed with the interaction of the cAMP and MAPK signaling pathways in the activation of lymphocytes. In this case, components of the cAMP pathway directly and indirectly affect MAPK signaling pathway meant to activate genes involving immunity and lymphocytes.
Newly formed cAMP is released from the membrane and diffuses across the intracellular space where it serves to activate PKA. The catalytic subunit of PKA must bind four molecules of cAMP to be activated, whereupon activation consists of cleavage between the regulatory and catalytic subunits.[4] This cleavage in turn activates PKA by exposing the catalytic sites of the C subunits, which can then phosphorylate an array of proteins in the cell.[4]
In lymphocytes, the intracellular levels of cAMP increase upon antigen-receptor stimulation and even more so in response to prostaglandin E and other immunosuppression agents.[8] In this case, cAMP serves to inhibit immunity players. PKA type I colocalizes with the T-cell and B-cell antigen receptors[9] and causes inhibition of T- and B-cell activation. PKA has even been highlighted as a direct inducer of genes contributing to immunosuppression.[10]
Additionally, the cAMP pathway also interacts with the MAPK pathway in a more indirect manner through its interaction with hematopoietic PTPase (HePTP). HePTP is expressed in all leukocytes. When overexpressed in T-cells, HePTP reduces the transcriptional activation of the interleukin-2 promoter typically induced by the activated T-cell receptor through a MAPK signaling cascade.[11] The way that HePTP effectively inhibits the MAPK signaling is by interacting with the MAP kinases Erk1, Erk2, and p38 through a short sequence in HePTP's non-catalytic N terminus termed the kinase interaction motif (KIM).,[11][12] The highly-specific binding of Erk and p38 to this subunit of HePTP results in rapid inactivation of the signaling cascade (see figure 3).
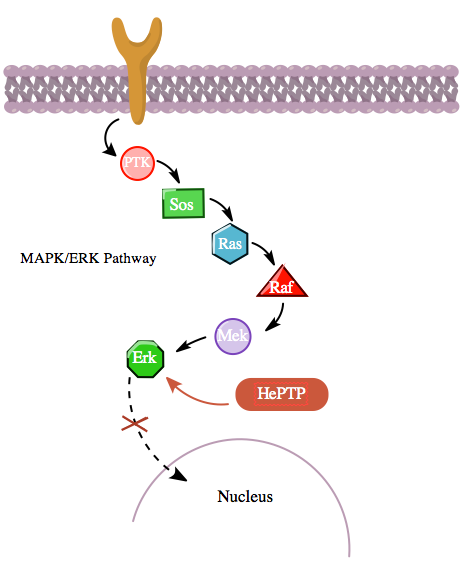
Yet, since both HePTP and Erk are cytosolic enzymes,[13] it is reasonable to conclude that there exists a mechanism for the inhibition of Erk by HePTP to cease in order to allow for the translocation of activated Erk to the nucleus. Indeed, like in many other cases of protein-protein interaction, HePTP appears to be phosphorylated by Erk and p38 at the sites Thr45 and Ser72.[11] Importantly though, a third phosphorylation site in the non-catalytic N terminus (the KIM region) of HePTP has been found—one that is phosphorylated to a much higher stoichiometry by the cAMP pathway,[1] in yet another instance of crosstalk between the cAMP and MAPK pathways.
Phosphorylation of this third site by PKAs from the cAMP pathway inhibits binding of MAP kinases to HePTP and thereby upregulates the MAPK/ERK signaling cascade. The MAPK pathway, through Ras, Raf, Mek, and Erk, shows low activity in the presence of unphosphorylated (active) HePTP. However, activation the cAMP pathway stimulates the activation of PKA, which in turn phosphorylates HePTP at Ser23. This prevents HePTP from binding to Erk and frees the MAPK pathway from inhibition, allowing downstream signaling to continue (see figure 4).
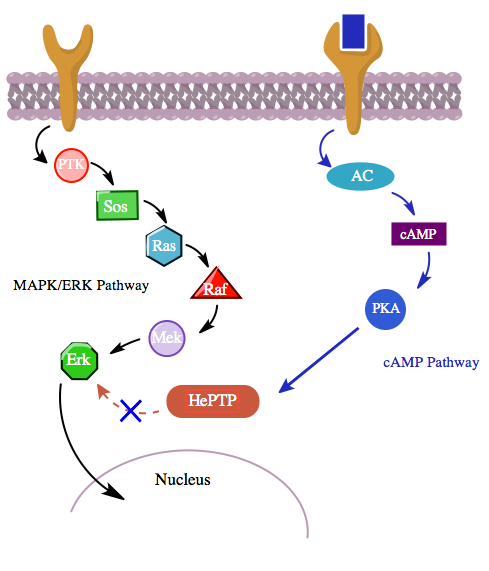
Moreover, studies involving smooth muscle cells from the atrium of the heart have shown that PKA can reduce the activation of MAP kinases in response to platelet-derived growth factor (PDGF) by phosphorylating the kinase c-Raf.[14] Thus, it seems plausible that PKA in the cAMP pathway could even be further involved in the regulation of lymphocyte activation not only by inhibiting the antigen-receptor MAPK signal pathway at its final stage, but even further upstream.
References
- Saxena, M. (1999), "Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase", Nat. Cell Biol. 1 (5): 305–311, doi:10.1038/13024, PMID 10559944 https://dx.doi.org/10.1038%2F13024
- Scott, J. D. (1991), "Cyclic nucleotide-dependent protein kinases", Pharmacol. Ther. 50 (1): 123–145, doi:10.1016/0163-7258(91)90075-W, PMID 1653962 https://dx.doi.org/10.1016%2F0163-7258%2891%2990075-W
- Krupinski J. (1989), "Adenylyl cyclase amino acid sequence: Possible channel- or transporter-like structure", Science 244 (4912): 1558–1564, doi:10.1126/science.2472670, PMID 2472670, Bibcode: 1989Sci...244.1558K https://dx.doi.org/10.1126%2Fscience.2472670
- Wine, Jeffrey. (1999–2008), "Across the Membrane; Intracellular Messengers: cAMP and cGMP", Stanford University, PSYCH121.
- Katz (2000), "Physical state of the extracellular matrix regulates the structure and molecular composition of cell–matrix adhesions", Mol. Biol. Cell 11 (3): 1047–1060, doi:10.1091/mbc.11.3.1047, PMID 10712519 http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=14830
- Philip J.S. Stork & John M. Schmitt. (2002), "Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation", Trends in Cell Biology 12 (6): 258–266, doi:10.1016/S0962-8924(02)02294-8, PMID 12074885, https://digitalcommons.georgefox.edu/cgi/viewcontent.cgi?article=1044&context=bio_fac
- Geiger, B. (2001), "Physical state of the extracellular matrix regulates the structure and molecular composition of cell–matrix adhesions", Nature Reviews Molecular Cell Biology 2 (11): 793–805, doi:10.1038/35099066, PMID 11715046 https://dx.doi.org/10.1038%2F35099066
- Ledbetter (1986), "Antibody binding to CD5 (Tp67) and Tp44 T cell surface molecules: effects on cyclic nucleotides, cytoplasmic free calcium, and cAMP-mediated suppression", Journal of Immunology 137 (10): 3299–3305, PMID 3021852 http://www.ncbi.nlm.nih.gov/pubmed/3021852
- Levy (1996), "Cyclic AMP-dependent protein kinase (cAK) in human B cells: co-localization of type I cAK (RIα2C2) with the antigen receptor during anti-immunoglobulin-induced B cell activation", Eur. J. Immunol. 26 (6): 1290–1296, doi:10.1002/eji.1830260617, PMID 8647207 https://dx.doi.org/10.1002%2Feji.1830260617
- Whisler (1991), "Cyclic AMP modulation of human B cell proliferative responses: role of cAMP-dependent protein kinases in enhancing B cell responses to phorboldiesters and ionomycin", Cell. Immunol. 142 (2): 398–415, doi:10.1016/0008-8749(92)90300-e, PMID 1320464 https://dx.doi.org/10.1016%2F0008-8749%2892%2990300-e
- Saxena, M. (1999), "Inhibition of T cell signaling by MAP kinase-targeted hematopoietic tyrosine phosphatase (HePTP)", J. Biol. Chem. 274 (17): 11693–700, doi:10.1074/jbc.274.17.11693, PMID 10206983 https://dx.doi.org/10.1074%2Fjbc.274.17.11693
- Pulido, R. (1998), "PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif", EMBO J. 17 (24): 7337–7350, doi:10.1093/emboj/17.24.7337, PMID 9857190 http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1171079
- Cobb (1994), "Regulation of the MAP kinase cascade", Cell. Mol. Biol. Res. 40 (3): 253–256, PMID 7874203 http://www.ncbi.nlm.nih.gov/pubmed/7874203
- Graves (1993), "Protein kinase A antagonizes platelet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells", Proc. Natl. Acad. Sci. U.S.A. 90 (21): 10300–10304, doi:10.1073/pnas.90.21.10300, PMID 7694289, Bibcode: 1993PNAS...9010300G http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=47762

