Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michela Castagna | + 2110 word(s) | 2110 | 2021-10-25 08:51:35 | | | |
| 2 | Camila Xu | Meta information modification | 2110 | 2021-11-09 07:19:06 | | | | |
| 3 | Paola Marciani | -1 word(s) | 2109 | 2021-11-09 15:46:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Castagna, M.; Algerta, M.; Galli, A.; Marciani, P.; Dule, N.; Perego, C. Iron-Homeostasis in Beta-Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/15817 (accessed on 13 July 2026).
Castagna M, Algerta M, Galli A, Marciani P, Dule N, Perego C. Iron-Homeostasis in Beta-Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/15817. Accessed July 13, 2026.
Castagna, Michela, Marku Algerta, Alessandra Galli, Paola Marciani, Nevia Dule, Carla Perego. "Iron-Homeostasis in Beta-Cells" Encyclopedia, https://encyclopedia.pub/entry/15817 (accessed July 13, 2026).
Castagna, M., Algerta, M., Galli, A., Marciani, P., Dule, N., & Perego, C. (2021, November 09). Iron-Homeostasis in Beta-Cells. In Encyclopedia. https://encyclopedia.pub/entry/15817
Castagna, Michela, et al. "Iron-Homeostasis in Beta-Cells." Encyclopedia. Web. 09 November, 2021.
Copy Citation
Iron is a central role in a variety of essential cellular functions as oxygen transport and exchange, being the metal component of many intracellular enzymes. Its ability to react with oxygen also makes it a toxic compound, able to generate reactive oxygen species (ROS) that can damage DNA, phospholipids and proteins. It is therefore of utmost importance, for both the cells and the organisms, to maintain iron homeostasis, ensuring iron supply and preventing accumulation of iron excess.
Iron metabolism
beta-cell function
plasma membrane
1. Introduction
Several disease states are characterised by aberrant iron handling. Abnormal iron homeostasis has been detected in hemochromatosis, anaemia, atherosclerosis, and in neurological diseases, such as Parkinson’s, Alzheimer’s, Huntington’s, Friedreich’s ataxia and the eating disorder pica [1][2][3][4][5][6][7][8][9][10][11].
Increasing evidence also points to a causal role of iron in diabetes. Iron is essential for insulin secretion [12][13], yet its accumulation is an important determinant of pancreatic islet inflammation and is considered a biomarker of diabetes risk and mortality [14].
The link between iron and diabetes first emerged considering pathological conditions as hemochromatosis and beta thalassemia [15][16][17][18], in which an involvement of iron overload in both beta-cell failure and insulin resistance was highlighted.
In addition, in type 2 diabetes mellitus (T2DM) subjects, increased levels of ferritin [19], a biomarker of increased body iron stores, and reduced levels of hepcidin, the hepatic hormone responsible for the systemic iron homeostasis, have been detected in the blood, highlighting the systemic alteration of iron metabolism [20].
2. Iron-Homeostasis in Beta-Cells
Due to its chemical nature and its possible harmful effects, cells have developed a complex system to handle iron: carriers and receptors bind and transport the ion across the membranes, enzymes and buffering proteins control its redox state and free level and iron regulatory proteins modulate the expression of iron-binding proteins, according to the ion level. Pancreatic beta-cells possess several of these proteins, although some specific players of this relevant process are not yet completely defined (Figure 1).
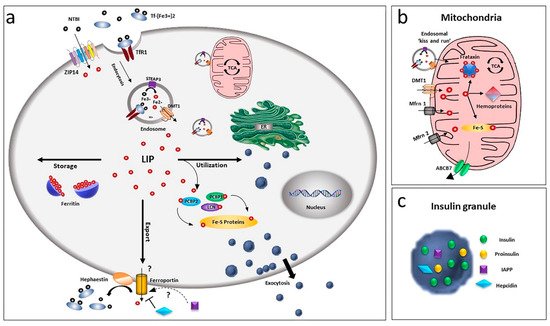
Figure 1. Overview of iron homeostasis in beta-cells. (a) Iron uptake in beta-cells is mediated by endocytosis of the transferrin-transferrin receptor complex and its release from endosomes by the divalent metal ion transporter DMT1. As non-transferrin-bound iron (NTBI), it can also be imported by the zinc transporter ZIP14 transporter. Being toxic as a free ion, Fe2+ is then readily distributed for storage, bound to ferritin, or for utilisation by chaperoning proteins as PCBPs and lipocalin. Iron efflux is mediated by ferroportin, a process regulated by hepcidin and hephaestin. LIP: labile iron pool. (b) Within the cell, the major site of utilisation is the mitochondria, where the ion is transported via DMT1 and mitoferrin (Mfrn1, Mfrn2) and inserted into heme and Fe/S cluster prosthetic groups. Mitochondria iron efflux is probably mediated by the ATP-binding cassette (ABC) transporter ABCB7. (c) Beta-cells, together with insulin, release IAPP and hepcidin, involved in a possible modulation of iron metabolism by an autocrine mechanism, via regulation of ferroportin.
2.1. Iron Influx through the Plasma Membrane
Uptake of iron in beta-cells is performed by two different systems: a receptor-mediated transport for the transferrin-bound iron (TBI) and a non-transferrin-bound iron (NTBI) transport. The first mechanism is based on the interaction of transferrin-bound iron with the specific cell surface transferrin receptor 1 (TfR1) [13][21]. The complex is then internalised in endocytic compartments in conjunction with the divalent metal ion transporter DMT1 (or SLC11A2) and the metalloreductase six transmembrane epithelial antigen of the prostate family member 3 (STEAP3) [22].
Internalised vesicles then fuse with lysosomal compartments, and the acidic milieu prompts the conformational change of Tf-Fe complex and the release of Fe3+, enabling its reduction to the ferrous form by STEAP3. Fe2+ is extruded in the cytoplasm through DMT1, exploiting the H+ gradient created by the vacuolar H+-ATPase (v-ATPase) as the driving force [23].
Recently, a non-transferrin-bound iron (NTBI) uptake has also been described in the human beta-cell line βlox5 [24]. The chemical nature of plasma NTBI is not known but is believed to mainly exist in ferric citrate and other low-molecular-weight species [25][26]. In some pathological conditions, higher molecular weight NTBI plasma fractions have been detected, suggesting the possible binding of Fe2+ and Fe3+ to proteins [26] and the existence of different NTBI pools, depending on the iron overload conditions [27]. NTBI can be observed in the blood of patients with iron overload conditions when transferrin is saturated [28], although its presence has also been detected at not fully saturated transferrin levels [29]. Interestingly, in diabetic subjects NTBI is already present at transferrin saturations below 60% [30]. In primary human islets, NTBI uptake is mediated by the zinc transporter ZIP14 (SLC39A14), which localises to the plasma membrane of beta-cells, where iron loading is restricted. Chronic (24 h) high glucose levels upregulate the transporter expression, thus confirming the functional relevance of ZIP14 and suggesting possible consequences in iron homeostasis [31]. However, siRNA-mediated ZIP14 knockdown determined only a 50% reduction of NTBI uptake, suggesting that other transport systems may be involved as well. A role of L-type or T-type calcium channels seems unlikely due to the lack of iron overload in murine beta-cells expressing them [32].
2.2. Iron Efflux through the Plasma Membrane
The exit of iron from beta-cells is controversial: ferroportin/Ireg1 (FPN1, SLC40A1) so far is the only known exporter for iron [33][34], and islets show a very low immunoreactivity for this transporter [35], although they express hephaestin. This protein is responsible for the membrane stabilisation of ferroportin and the oxidation of Fe2+ to Fe3+ required for the interaction with transferrin [35].
Interestingly, beta-cells, together with insulin, also release hepcidin that is known to bind ferroportin and induce its internalisation [36][37], thus suggesting a positive feedback mechanism in iron regulation during glucose-stimulated insulin secretion, mediated by ferroportin control [38].
Another possible modulator of ferroportin is the islet amyloid polypeptide (IAPP) [39], which is released together with insulin and plays a role in glucose homeostasis [40] and in the control of food intake [41][42]. Although its role in iron homeostasis in beta-cells has not yet been established, it could suggest a parallelism with neurons, in which the amyloid polypeptide APP stabilises ferroportin at the plasma membrane and stimulates iron release through ferroxidase activity [43][44][45][46][47][48], thus preventing iron overload and oxidative stress.
2.3. Iron Binding Proteins
By a tight control of iron homeostasis, cells avoid excess of harmful free iron. Once inside the cell, iron forming the cytoplasmic labile pool (LIP) is sequestered by ferritin, the exclusive cytosolic iron-storage protein. Both H and L chains are expressed in beta-cells and modulated at the translational level by iron overload: when iron increases, ferritin synthesis increases as well as iron storage [49]. By sequestering the element, ferritin play a role in iron detoxification and functions as an iron reserve protein. Although the presence of a cytoplasmic labile iron pool consisting of chelatable iron has been detected in the past, concerns have been raised that iron, once internalised in cells, is delivered to ferritin via direct protein-protein interactions in a hydrophobic microenvironment, since LIP does not seem to have the chemical characteristics of an intermediate iron pool [50]. Chaperone proteins, such as poly r(C)-binding proteins (PCBPs) [51], are involved in this process.
All four known PCBP isoforms can bind and deliver iron to the cytosolic ferritin [49][52], but they show different abilities as iron chaperones. For example, only PCBP2 can bind to the carrier systems DMT1 and FPN1 in an iron-dependent way [53][54]. Both PCBP1 and PCBP2 can deliver iron to ferritin, but only PCBP1 is fundamental in ferritinophagy, an iron recycling process [55] in which the iron-ferritin complex is captured by the nuclear receptor coactivator-4 (NCOA4) and directed into the autophagosome [56].
The expression of both PCBP1 and PCBP2 has been documented in beta-cells, but their specific role in iron handling and whether they are also involved in iron delivery to intracellular organelles and Fe-S proteins remains to be elucidated in this cell type.
2.4. Iron Exchange with Organelles
Although iron has been detected in almost all intracellular organelles, mitochondria are the main station of cellular iron metabolism. They are indeed a site of iron storage and utilisation. Vital synthesis of heme and iron-sulphur (Fe-S) clusters for electron transport proteins take place within them.
The iron exchange with mitochondria is thought to be mediated by DMT1 and the classical mitochondrial iron transporters mitoferrin (Mfrn) 1 and 2 [57], being the second more specific for non-erythroid cells [58]. Lipocalin (LCN) protein 2 is also involved in this process as a chaperon protein [13][59]. In HEK293 cells, permanently expressing DMT1, the transporter is present at the outer mitochondrial membrane (OMM) [60] and found to be involved in Fe2+ and Mn2+ uptake [61]. Mfrn1 and 2 ensure the iron transport across the inner mitochondrial membrane, where the element is utilised for heme synthesis and Fe-S clusters biogenesis or is sequestered by mitochondrial ferritin (MTFT).
Fe-S cluster biogenesis requires frataxin, an iron mitochondrial chaperone expressed in islets and beta-cells and stimulated by hyperglycaemic conditions [62]. Individuals affected by Friedreich’s ataxia (FRDA), a neurodegenerative disorder caused by frataxin deficiency, also develop non-neurological symptoms, such as diabetes or glucose intolerance (8 to 32% incidence) [63]. In these patients, iron overload and increased beta-cell apoptosis have been observed, thus further supporting a link between iron dyshomeostasis and diabetes.
Considering the exit of iron from the mitochondrial matrix, the ATP-binding cassette (ABC) transporter ABCB7 is believed to export iron in the form of Fe-S clusters. This hypothesis is based on the activity of the yeast orthologue Atm1 [64] that can transport glutathione-coordinated Fe-S clusters, connecting the mitochondrial and cytosolic Fe-S cluster assembly systems [65][66]. Recently, Pearson et al. confirmed this substrate specificity, highlighting the role of Mg-ATP in the transport process [67]. An additional mechanism for the exit of iron from the mitochondrial matrix could be the export of heme by specific transporters [68].
Iron can also be delivered to mitochondria by direct communication with other organelles. In developing erythroid cells, requiring a very efficient delivery of iron to mitochondria for heme synthesis, a direct delivery of iron from endosomes to mitochondria by a “kiss and run” mechanism [69]) has also been described, in which the transfer of the cation would be mediated by the docking of mitochondria and transferrin-loaded endosomes through the voltage-dependent anion channel 1 (VDAC1) or DMT1 [61][70]. Due to the relevant role played by iron in insulin release (see below), similar mechanisms of iron delivery could also be envisaged for beta-cells, considering that the same process has been described in epithelial cells [71].
Contact sites between mitochondria and lysosomes, not related to mitophagy or lysosomal degradation of mitochondrial vesicles, have also been described by high-resolution microscopy [72]. Supporting the functional relevance of such a contact in iron transport, in erythroid progenitors, where the TfR2 isoform mediates the delivery of lysosomal transferrin to mitochondria, TfR2 deficiency reduced mitochondrial size and heme production [73]. Furthermore, in fibroblasts of patients affected by neurodegeneration with brain iron accumulation, mitochondrial function abnormalities and reduced lysosomal proteolytic activity have been observed [74], suggesting a further mechanism of intracellular iron trafficking based on the interaction between mitochondria and lysosomes.
Mitochondrial-associated ER membranes (MAMs) could be also implicated in cell iron homeostasis. Deficiency of Cisd2 (CDGSH iron sulphur domain 2), an Fe-S protein localised on MAMs, leads to mitochondrial dysfunction and disturbance of intracellular Ca2+ homeostasis, resulting in insulin insensitivity in adipocytes [75]. Interestingly, in yeast, loss of the protein complex ERMES (endoplasmic reticulum mitochondria encounter structure) connecting the two organelles, determines an iron-deficiency response even in iron-repleted conditions, causing iron excess in the cell [76]. Furthermore, dominant mutants of the vacuolar protein sorting 13 (VSP13p) rescue ERMES mutants, suppressing the iron deficiency response. No transporters for the delivery of iron to endoplasmic reticulum (ER) have been identified so far. The 2Fe-2S protein iron sulphur domain 2 (Miner 1) that localises to ER in other cell types and is relevant for ER integrity [77][78] could be involved in this function [79].
2.5. Iron Metabolism Regulatory Proteins
As both iron deficiency and overload can be detrimental, in beta-cells, iron-genes are post-transcriptionally regulated by the iron regulatory proteins (IRPs), based on iron availability [80][81][82]. These are RNA-binding proteins that, by binding to IRE sequences present on mRNAs of iron handling proteins, modulate their translation. In particular, in conditions of iron deficiency, IRP binds to TfR1, DMT1, and ferritin mRNAs and promotes their translation, thus increasing cellular iron absorption and iron storage [80]. At the same time, IRPs suppress FPN1 translation, thus reducing cellular iron release [83]. Both IRP1 and IRP2 are expressed in beta-cells, and IRP2 knockout mice develop diabetes due to misregulation of iron metabolism. Pro-insulin translational fidelity in pancreatic beta-cells requires the activity of the Fe-S cluster enzyme CDKAL1. This enzyme is responsible for the adenosine methylthiolation in the tRNA for lysine, a modification necessary to maintain the accuracy of codon recognition during protein translation. CDKAL1 dysfunction causes a misreading of the codon and impaired proinsulin processing and release. Mice lacking IRP2 protein develop diabetes because the consequent iron deficiency leads to a reduced function of CDKAL1 [84].
References
- Nevo, Y.; Nelson, N. The NRAMP Family of Metal-Ion Transporters. Biochim. Biophys. Acta BBA Mol. Cell Res. 2006, 1763, 609–620.
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 Is Mutated in the Anemic Belgrade (b) Rat: Evidence of a Role for Nramp2 in Endosomal Iron Transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153.
- Herrmann, T.; Muckenthaler, M.; van der Hoeven, F.; Brennan, K.; Gehrke, S.G.; Hubert, N.; Sergi, C.; Gröne, H.-J.; Kaiser, I.; Gosch, I.; et al. Iron Overload in Adult Hfe-Deficient Mice Independent of Changes in the Steady-State Expression of the Duodenal Iron Transporters DMT1 and Ireg1/Ferroportin. J. Mol. Med. 2004, 82, 39–48.
- Muckenthaler, M.; Roy, C.N.; Custodio, A.O.; Miñana, B.; deGraaf, J.; Montross, L.K.; Andrews, N.C.; Hentze, M.W. Regulatory Defects in Liver and Intestine Implicate Abnormal Hepcidin and Cybrd1 Expression in Mouse Hemochromatosis. Nat. Genet. 2003, 34, 102–107.
- Fergelot, P.; Orhant, M.; Thénié, A.; Loyer, P.; Ropert-Bouchet, M.; Lohyer, S.; Le Gall, J.-Y.; Mosser, J. Over-Expression of Wild-Type and Mutant HFE in a Human Melanocytic Cell Line Reveals an Intracellular Bridge between MHC Class I Pathway and Transferrin Iron Uptake. Biol. Cell 2003, 95, 243–255.
- Li, W.; Hellsten, A.; Nyhalah, J.D.; Yuan, X.-M. Enhanced Expression of Natural Resistance-Associated Macrophage Protein 1 in Atherosclerotic Lesions May Be Associated with Oxidized Lipid-Induced Apoptosis. Ann. N. Y. Acad. Sci. 2004, 1030, 202–207.
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of Mitochondrial Iron Accumulation by Yfh1p, a Putative Homolog of Frataxin. Science 1997, 276, 1709–1712.
- Andrews, N.C.; Levy, J.E. Iron Is Hot: An Update on the Pathophysiology of Hemochromatosis. Blood 1998, 92, 1845–1851.
- Askwith, C.; Kaplan, J. Iron and Copper Transport in Yeast and Its Relevance to Human Disease. Trends Biochem. Sci. 1998, 23, 135–138.
- Moos, T.; Morgan, E.H. The Metabolism of Neuronal Iron and Its Pathogenic Role in Neurological Disease: Review. Ann. N. Y. Acad. Sci. 2004, 1012, 14–26.
- Borgna-Pignatti, C.; Zanella, S. Pica as a Manifestation of Iron Deficiency. Expert Rev. Hematol. 2016, 9, 1075–1080.
- Nielsen, J.H. Mechanisms of Pancreatic Beta-Cell Growth and Regeneration: Studies on Rat Insulinoma Cells. Exp. Clin. Endocrinol. 1989, 93, 277–285.
- Hansen, J.B.; Tonnesen, M.F.; Madsen, A.N.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.; Heller, R.S.; Nielsen, A.Ø.; Størling, J.; Baeyens, L.; et al. Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic β Cell Fate in Response to Cytokines. Cell Metab. 2012, 16, 449–461.
- Simcox, J.A.; McClain, D.A. Iron and Diabetes Risk. Cell Metab. 2013, 17, 329–341.
- Buysschaert, M.; Paris, I.; Selvais, P.; Hermans, M.P. Clinical Aspects of Diabetes Secondary to Idiopathic Haemochromatosis in French-Speaking Belgium. Diabetes Metab. 1997, 23, 308–313.
- Moirand, R.; Adams, P.C.; Bicheler, V.; Brissot, P.; Deugnier, Y. Clinical Features of Genetic Hemochromatosis in Women Compared with Men. Ann. Intern. Med. 1997, 127, 105–110.
- Dmochowski, K.; Finegood, D.T.; Francombe, W.; Tyler, B.; Zinman, B. Factors Determining Glucose Tolerance in Patients with Thalassemia Major. J. Clin. Endocrinol. Metab. 1993, 77, 478–483.
- Merkel, P.A.; Simonson, D.C.; Amiel, S.A.; Plewe, G.; Sherwin, R.S.; Pearson, H.A.; Tamborlane, W.V. Insulin Resistance and Hyperinsulinemia in Patients with Thalassemia Major Treated by Hypertransfusion. N. Engl. J. Med. 1988, 318, 809–814.
- Rajpathak, S.N.; Crandall, J.P.; Wylie-Rosett, J.; Kabat, G.C.; Rohan, T.E.; Hu, F.B. The Role of Iron in Type 2 Diabetes in Humans. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 671–681.
- Altamura, S.; Kopf, S.; Schmidt, J.; Müdder, K.; da Silva, A.R.; Nawroth, P.; Muckenthaler, M.U. Uncoupled Iron Homeostasis in Type 2 Diabetes Mellitus. J. Mol. Med. 2017, 95, 1387–1398.
- Lu, J.P.; Hayashi, K.; Awai, M. Transferrin Receptor Expression in Normal, Iron-Deficient and Iron-Overloaded Rats. Pathol. Int. 1989, 39, 759–764.
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The Long History of Iron in the Universe and in Health and Disease. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 161–187.
- Ohta, T.; Yamamoto, M.; Numata, M.; Iseki, S.; Kitagawa, H.; Kayahara, M.; Nagakawa, T.; Miwa, K.; Nakagawa, A.; Morise, T.; et al. Differential Expression of Vacuolar-Type H+-ATPase between Normal Human Pancreatic Islet B-Cells and Insulinoma Cells. Int. J. Oncol. 1997, 11, 597–601.
- Coffey, R.; Knutson, M.D. The Plasma Membrane Metal-Ion Transporter ZIP14 Contributes to Nontransferrin-Bound Iron Uptake by Human β-Cells. Am. J. Physiol. Cell Physiol. 2017, 312, C169–C175.
- Grootveld, M.; Bell, J.D.; Halliwell, B.; Aruoma, O.I.; Bomford, A.; Sadler, P.J. Non-Transferrin-Bound Iron in Plasma or Serum from Patients with Idiopathic Hemochromatosis. Characterization by High Performance Liquid Chromatography and Nuclear Magnetic Resonance Spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422.
- Evans, R.W.; Rafique, R.; Zarea, A.; Rapisarda, C.; Cammack, R.; Evans, P.J.; Porter, J.B.; Hider, R.C. Nature of Non-Transferrin-Bound Iron: Studies on Iron Citrate Complexes and Thalassemic Sera. J. Biol. Inorg. Chem. 2008, 13, 57–74.
- Porter, J.B.; Walter, P.B.; Neumayr, L.D.; Evans, P.; Bansal, S.; Garbowski, M.; Weyhmiller, M.G.; Harmatz, P.R.; Wood, J.C.; Miller, J.L.; et al. Mechanisms of Plasma Non-Transferrin Bound Iron Generation: Insights from Comparing Transfused Diamond Blackfan Anaemia with Sickle Cell and Thalassaemia Patients. Br. J. Haematol. 2014, 167, 692–696.
- Hershko, C.; Graham, G.; Bates, G.W.; Rachmilewitz, E.A. Non-Specific Serum Iron in Thalassaemia: An Abnormal Serum Iron Fraction of Potential Toxicity. Br. J. Haematol. 1978, 40, 255–263.
- Loréal, O.; Gosriwatana, I.; Guyader, D.; Porter, J.; Brissot, P.; Hider, R.C. Determination of Non-Transferrin-Bound Iron in Genetic Hemochromatosis Using a New HPLC-Based Method. J. Hepatol. 2000, 32, 727–733.
- Lee, D.-H.; Liu, D.Y.; Jacobs, D.R.; Shin, H.-R.; Song, K.; Lee, I.-K.; Kim, B.; Hider, R.C. Common Presence of Non-Transferrin-Bound Iron among Patients with Type 2 Diabetes. Diabetes Care 2006, 29, 1090–1095.
- Maxel, T.; Smidt, K.; Petersen, C.C.; Honoré, B.; Christensen, A.K.; Jeppesen, P.B.; Brock, B.; Rungby, J.; Palmfeldt, J.; Larsen, A. The Zinc Transporter Zip14 (SLC39a14) Affects Beta-Cell Function: Proteomics, Gene Expression, and Insulin Secretion Studies in INS-1E Cells. Sci. Rep. 2019, 9, 8589.
- Schulla, V. Impaired Insulin Secretion and Glucose Tolerance in Cell-Selective CaV1.2 Ca2+ Channel Null Mice. EMBO J. 2003, 22, 3844–3854.
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional Cloning of Zebrafish Ferroportin1 Identifies a Conserved Vertebrate Iron Exporter. Nature 2000, 403, 776–781.
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A Novel Duodenal Iron-Regulated Transporter, IREG1, Implicated in the Basolateral Transfer of Iron to the Circulation. Mol. Cell 2000, 5, 299–309.
- Hudson, D.M.; Curtis, S.B.; Smith, V.C.; Griffiths, T.A.M.; Wong, A.Y.K.; Scudamore, C.H.; Buchan, A.M.J.; MacGillivray, R.T.A. Human Hephaestin Expression Is Not Limited to Enterocytes of the Gastrointestinal Tract but Is Also Found in the Antrum, the Enteric Nervous System, and Pancreatic β-Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G425–G432.
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093.
- Kulaksiz, H.; Fein, E.; Redecker, P.; Stremmel, W.; Adler, G.; Cetin, Y. Pancreatic Beta-Cells Express Hepcidin, an Iron-Uptake Regulatory Peptide. J. Endocrinol. 2008, 197, 241–249.
- Aigner, E.; Felder, T.K.; Oberkofler, H.; Hahne, P.; Auer, S.; Soyal, S.; Stadlmayr, A.; Schwenoha, K.; Pirich, C.; Hengster, P.; et al. Glucose Acts as a Regulator of Serum Iron by Increasing Serum Hepcidin Concentrations. J. Nutr. Biochem. 2013, 24, 112–117.
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J.; Porte, D. Evidence of Cosecretion of Islet Amyloid Polypeptide and Insulin by Beta-Cells. Diabetes 1990, 39, 634–638.
- Ritzel, R.A.; Meier, J.J.; Lin, C.-Y.; Veldhuis, J.D.; Butler, P.C. Human Islet Amyloid Polypeptide Oligomers Disrupt Cell Coupling, Induce Apoptosis, and Impair Insulin Secretion in Isolated Human Islets. Diabetes 2007, 56, 65–71.
- Lutz, T.A.; Del Prete, E.; Scharrer, E. Reduction of Food Intake in Rats by Intraperitoneal Injection of Low Doses of Amylin. Physiol. Behav. 1994, 55, 891–895.
- Morley, J.E.; Flood, J.F.; Horowitz, M.; Morley, P.M.; Walter, M.J. Modulation of Food Intake by Peripherally Administered Amylin. Am. J. Physiol. 1994, 267, R178–R184.
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-Export Ferroxidase Activity of β-Amyloid Precursor Protein Is Inhibited by Zinc in Alzheimer’s Disease. Cell 2010, 142, 857–867.
- McCarthy, R.C.; Park, Y.; Kosman, D.J. sAPP Modulates Iron Efflux from Brain Microvascular Endothelial Cells by Stabilizing the Ferrous Iron Exporter Ferroportin. EMBO Rep. 2014, 15, 809–815.
- Rogers, J.T.; Venkataramani, V.; Washburn, C.; Liu, Y.; Tummala, V.; Jiang, H.; Smith, A.; Cahill, C.M. A Role for Amyloid Precursor Protein Translation to Restore Iron Homeostasis and Ameliorate Lead (Pb) Neurotoxicity. J. Neurochem. 2016, 138, 479–494.
- Dlouhy, A.C.; Bailey, D.K.; Steimle, B.L.; Parker, H.V.; Kosman, D.J. Fluorescence Resonance Energy Transfer Links Membrane Ferroportin, Hephaestin but Not Ferroportin, Amyloid Precursor Protein Complex with Iron Efflux. J. Biol. Chem. 2019, 294, 4202–4214.
- Tsatsanis, A.; Dickens, S.; Kwok, J.C.F.; Wong, B.X.; Duce, J.A. Post Translational Modulation of β-Amyloid Precursor Protein Trafficking to the Cell Surface Alters Neuronal Iron Homeostasis. Neurochem. Res. 2019, 44, 1367–1374.
- Tsatsanis, A.; Wong, B.X.; Gunn, A.P.; Ayton, S.; Bush, A.I.; Devos, D.; Duce, J.A. Amyloidogenic Processing of Alzheimer’s Disease β-Amyloid Precursor Protein Induces Cellular Iron Retention. Mol. Psychiatry 2020, 25, 1958–1966.
- MacDonald, M.J.; Cook, J.D.; Epstein, M.L.; Flowers, C.H. Large Amount of (Apo)Ferritin in the Pancreatic Insulin Cell and Its Stimulation by Glucose. FASEB J. 1994, 8, 777–781.
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The New Role of Poly (RC)-Binding Proteins as Iron Transport Chaperones: Proteins That Could Couple with Inter-Organelle Interactions to Safely Traffic Iron. Biochim. Biophys. Acta BBA Gen. Subj. 2020, 1864, 129685.
- Philpott, C.C.; Jadhav, S. The Ins and Outs of Iron: Escorting Iron through the Mammalian Cytosol. Free Radic. Biol. Med. 2019, 133, 112–117.
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802.
- Yanatori, I.; Richardson, D.R.; Imada, K.; Kishi, F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J. Biol. Chem. 2016, 291, 17303–17318.
- Yanatori, I.; Kishi, F. DMT1 and Iron Transport. Free Radic. Biol. Med. 2019, 133, 55–63.
- Ryu, M.-S.; Duck, K.A.; Philpott, C.C. Ferritin Iron Regulators, PCBP1 and NCOA4, Respond to Cellular Iron Status in Developing Red Cells. Blood Cells Mol. Dis. 2018, 69, 75–81.
- Ryu, M.-S.; Zhang, D.; Protchenko, O.; Shakoury-Elizeh, M.; Philpott, C.C. PCBP1 and NCOA4 Regulate Erythroid Iron Storage and Heme Biosynthesis. J. Clin. Investig. 2017, 127, 1786–1797.
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016.
- Rouault, T.A. Mitochondrial Iron Overload: Causes and Consequences. Curr. Opin. Genet. Dev. 2016, 38, 31–37.
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 2014, 509, 105–109.
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for Mitochondrial Localization of Divalent Metal Transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145.
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A Role for Divalent Metal Transporter (DMT1) in Mitochondrial Uptake of Iron and Manganese. Sci. Rep. 2018, 8, 211.
- Del Guerra, S.; D’Aleo, V.; Gualtierotti, G.; Pandolfi, R.; Boggi, U.; Vistoli, F.; Barnini, S.; Filipponi, F.; Del Prato, S.; Lupi, R. Evidence for a Role of Frataxin in Pancreatic Islets Isolated from Multi-Organ Donors with and without Type 2 Diabetes Mellitus. Horm. Metab. Res. 2012, 44, 471–475.
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. In Brain Iron Metabolism and CNS Diseases; Chang, Y.-Z., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1173, pp. 125–143. ISBN 9789811395888.
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a Putative Mitochondrial Iron Transporter Gene (ABC7) in X-Linked Sideroblastic Anemia and Ataxia (XLSA/A). Hum. Mol. Genet. 1999, 8, 743–749.
- Bottomley, S.S.; May, B.K.; Cox, T.C.; Cotter, P.D.; Bishop, D.F. Molecular Defects of Erythroid 5-Aminolevulinate Synthase in X-Linked Sideroblastic Anemia. J. Bioenerg. Biomembr. 1995, 27, 161–168.
- Bottomley, S.S.; Fleming, M.D. Sideroblastic Anemia: Diagnosis and Management. Hematol. Oncol. Clin. N. Am. 2014, 28, 653–670.
- Pearson, S.A.; Wachnowsky, C.; Cowan, J.A. Defining the Mechanism of the Mitochondrial Atm1p Cluster Exporter. Metallomics 2020, 12, 902–915.
- May, A.; Bishop, D.F. The Molecular Biology and Pyridoxine Responsiveness of X-Linked Sideroblastic Anaemia. Haematologica 1998, 83, 56–70.
- Sheftel, A.D.; Zhang, A.-S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct Interorganellar Transfer of Iron from Endosome to Mitochondrion. Blood 2007, 110, 125–132.
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria–Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513.
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-Mitochondria Interactions Are Modulated by Iron Release from Transferrin. J. Cell Biol. 2016, 214, 831–845.
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-Lysosome Contacts Regulate Mitochondrial Fission via RAB7 GTP Hydrolysis. Nature 2018, 554, 382–386.
- Khalil, S.; Holy, M.; Grado, S.; Fleming, R.; Kurita, R.; Nakamura, Y.; Goldfarb, A. A Specialized Pathway for Erythroid Iron Delivery through Lysosomal Trafficking of Transferrin Receptor 2. Blood Adv. 2017, 1, 1181–1194.
- Seibler, P.; Burbulla, L.F.; Dulovic, M.; Zittel, S.; Heine, J.; Schmidt, T.; Rudolph, F.; Westenberger, A.; Rakovic, A.; Münchau, A.; et al. Iron Overload Is Accompanied by Mitochondrial and Lysosomal Dysfunction in WDR45 Mutant Cells. Brain 2018, 141, 3052–3064.
- Wang, C.-H.; Tsai, T.-F.; Wei, Y.-H. Role of Mitochondrial Dysfunction and Dysregulation of Ca(2+) Homeostasis in Insulin Insensitivity of Mammalian Cells. Ann. N. Y. Acad. Sci. 2015, 1350, 66–76.
- Xue, Y.; Schmollinger, S.; Attar, N.; Campos, O.A.; Vogelauer, M.; Carey, M.F.; Merchant, S.S.; Kurdistani, S.K. Endoplasmic Reticulum-Mitochondria Junction Is Required for Iron Homeostasis. J. Biol. Chem. 2017, 292, 13197–13204.
- Conlan, A.R.; Axelrod, H.L.; Cohen, A.E.; Abresch, E.C.; Zuris, J.; Yee, D.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L. Crystal Structure of Miner1: The Redox-Active 2Fe-2S Protein Causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153.
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.-F.; Tsai, T.-F.; Simon, M.I.; et al. Wolfram Syndrome Protein, Miner1, Regulates Sulphydryl Redox Status, the Unfolded Protein Response, and Ca2+ Homeostasis. EMBO Mol. Med. 2013, 5, 904–918.
- Backe, M.B.; Moen, I.W.; Ellervik, C.; Hansen, J.B.; Mandrup-Poulsen, T. Iron Regulation of Pancreatic Beta-Cell Functions and Oxidative Stress. Annu. Rev. Nutr. 2016, 36, 241–273.
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414.
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38.
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian Iron Metabolism and Its Control by Iron Regulatory Proteins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 1468–1483.
- Lymboussaki, A.; Pignatti, E.; Montosi, G.; Garuti, C.; Haile, D.J.; Pietrangelo, A. The Role of the Iron Responsive Element in the Control of Ferroportin1/IREG1/MTP1 Gene Expression. J. Hepatol. 2003, 39, 710–715.
- Dos Santos, M.C.F.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 Regulates Insulin Production through Iron-Mediated Cdkal1-Catalyzed TRNA Modification. Nat. Commun. 2020, 11, 296.
More
Information
Subjects:
Endocrinology & Metabolism
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
09 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No