 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmen Espinós | + 2539 word(s) | 2539 | 2020-11-05 05:13:51 | | | |
| 2 | Dean Liu | Meta information modification | 2539 | 2021-09-14 10:48:24 | | | | |
| 3 | Conner Chen | Meta information modification | 2539 | 2021-09-22 03:43:36 | | |
Video Upload Options
The syndromes of neurodegeneration with brain iron accumulation (NBIA) encompass a group of invalidating and progressive rare diseases that share the abnormal accumulation of iron in the basal ganglia. The onset of NBIA disorders ranges from infancy to adulthood. Main clinical signs are related to extrapyramidal features (dystonia, parkinsonism and choreoathetosis), and neuropsychiatric abnormalities. Ten NBIA forms are widely accepted to be caused by mutations in the genes PANK2, PLA2G6, WDR45, C19ORF12, FA2H, ATP13A2, COASY, FTL1, CP, and DCAF17. However, many patients remain without a genetic diagnosis, and therefore, there must be additional yet undiscovered NBIA genes. The genetic heterogeneity and the corresponding encoded proteins emphasize that several pathways are involved in NBIA syndromes: iron and lipid metabolism, mitochondrial dynamics, and autophagy. Moreover, for these forms as well as for many neurodegenerative conditions, mitochondrial dysfunction and oxidative stress are common mechanisms of disease.
1. Overview of the NBIA Syndromes
Ten genes are classically accepted as NBIA genes[1].The two major forms are PKAN (pantothenate kinase-associated neurodegeneration; 35–50%; PANK2 gene) and PLAN (PLA2G6-associated neurodegeneration; ~20%; PLA2G6 gene), followed by MPAN (mitochondrial membrane protein-associated neurodegeneration; 6–10%; C19ORF12 gene) and BPAN (β-propeller-associated neurodegeneration; 1–2%; WDR45 gene). FAHN (fatty acid hydroxylase-associated neurodegeneration; FA2H gene), NF (neuroferrinopathy; FTL gene), aceruloplasminemia (CP gene) and Woodhouse–Sakati syndrome (DCAF17 gene) are rare types. Finally, two probands and five probands, respectively, are described for Kufor–Rakeb syndrome (ATP13A2 gene) and CoPAN (COASY protein-associated neurodegeneration; COASY gene). Importantly, a relevant number of patients with NBIA have no genetic diagnosis, suggesting that other implicated genes remain to be discovered.
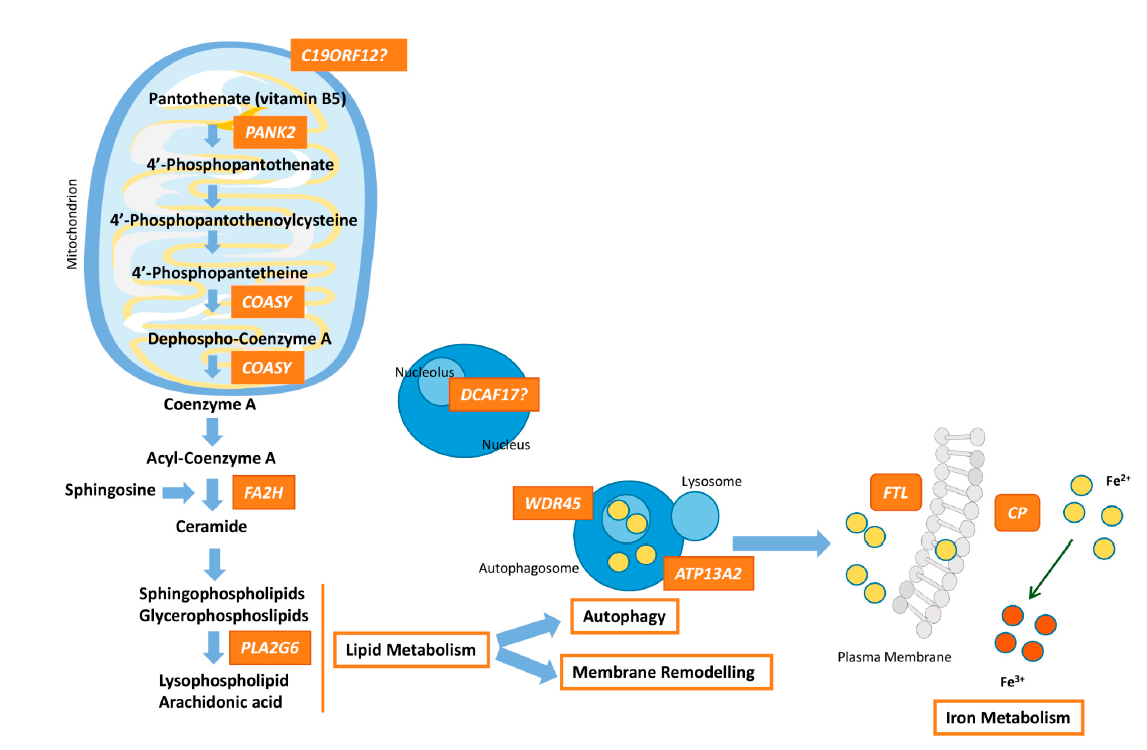
Figure 1. Pathway connecting lipid synthesis, lysosomes dysfunction and iron accumulation in NBIA syndromes. PANK2, COASY, FA2H and PLA2G6 defects lead to harmed phospholipid membrane synthesis and impaired myelination or myelin maintenance. Dysfunctional membranes may lead to lysosomal and mitochondrial damage causing ROS production and iron uptake upregulation. WDR45 and ATP13A2 are involved in autophagosome formation/degradation. The decrease of WDR45 protein expression causes the accumulation of aberrant autophagic structures. Lysosomal dysfunction due to ATP13A2 defect, which may originate from impaired phospholipid recycling, could ultimately cause degradation of substrates and damage of autophagosome clearance. Moreover, perturbation of lysosomes is important for iron homeostasis and may promote deposits of iron. Dysfunction of FTL prevents the recruitment of iron excess and malfunctioning of CP causes impairment of iron export from the cell, responsible for iron overload. The higher free iron-dependent oxidative damage requires that the cell increases the degradation of oxidized molecules, which can a ect the cellular recycling systems, such as lysosomes, and finally, lead to cellular death. The role of DCAF17 remains unclear, although its dysfunction leads to a neurodegenerative disorders, and hence, in some way, DCAF17 may be related to mitochondrial dynamics.
2. NBIA Errors of Coenzyme a Biosynthesis
2.1. Pantothenate Kinase-Associated Neurodegeneration (PKAN)
PANK2 encodes for the mitochondrial isoform of pantothenate kinase that catalyzes the ATP-dependent phosphorylation of pantothenate, an essential regulatory step in CoA biosynthesis (Figure 1). The PANK2 protein, located in mitochondria, has two domains: one that includes two MLS (mitochondrial localization signals) in the NH2-terminal and the other large one that encodes for the catalytic core of the enzyme[2][3]. To date, more than 120 disease-causing mutations are known in PANK2 (HGMD® Professional 2020.1; accessed 8 October 2020).
Two main subtypes are associated with the autosomal recessive (AR) disorder PKAN: classical (early onset, usually before 6 years of age) or atypical (first symptoms in early adulthood). The classical presentation is characterized by dystonic tremor with predominant oromandibular involvement, optic atrophy, pigmentary retinopathy, acanthocytosis, and spasticity. In the late-onset PKAN form, motor involvement tends to be less severe but cognitive decline and psychiatric alterations are predominant traits[4][5]. In PKAN patients, the characteristic “eye of tiger sign” is detected by T2-weighted magnetic resonance, which reflects the focal accumulation of iron in the globus pallidus (GP)[6][7]. The iron accumulation correlates with neural damage, often associated with neuroaxonal spheroids that represent degenerating neurons in GP[8][2].
In addition to this, neuronal degeneration promotes a mild inflammatory response due to infiltrations of iron-containing macrophages, astrogliosis and microglial activation[9]. These findings suggest that chronic neuronal hypoxia and/or ischemia in the GP may play an important role in the pathomechanism of PKAN.
2.2. COASY Protein-Associated Neurodegeneration (CoPAN)
CoA synthase, a bifunctional enzyme that catalyzes the last two steps in CoA biosynthesis, is encoded by the COASY gene (Figure 1). The protein contains a mitochondrial localization signal, a regulatory region and two domains for its catalytic kinase activities, 40PP adenyltransferase (PPAT) and dephospho-CoA kinase (DPCK), and it is predominantly located at the mitochondrial matrix. Mutations in this gene lead to CoPAN, an ultra-rare NBIA form inherited in an AR manner. Hitherto, four CoPAN families are known[10][11] and three missense variants have been described (HGMD® Professional 2020.1; accessed 10 September 2020). All these patients show typical NBIA features: onset in the first decade of life with mild cognitive impairment and gait difficulties, progressing to a more severe phenotype including dystonia, parkinsonism, dysarthria, spasticity and axonal neuropathy. Obsessive-compulsive disorder is also a common featurein CoPAN patients.
3. NBIA Types Related to Lipid Metabolism and Membrane Remodeling
3.1. PLA2G6-Associated Neurodegeneration (PLAN)
AR mutations in the phospholipase A2 group (PLA2G6) are causative of the PLAN phenotypic spectrum, including classic infantile neuronal dystrophy (INAD), atypical neuronal dystrophy (NAD) with childhood-onset, and an adult onset dystonia-parkinsonism form named PARK14[12]. This NBIA type is characterized by progressive motor deterioration and may lead to spastic or hypotonic tetraparesis with truncal hypotonia, cerebellar ataxia, early optic atrophy, seizures in later stages of the disease, dystonia, and cognitive impairment[13]. The most typical sign of INAD is a fast progression of cerebellar atrophy in early stages. At later stages of the disease progression, most INAD patients usually show brain iron accumulation in the GP and the substantia nigra[14][15]. About 200 distinct mutations in PLA2G6 of all types (missense, deletions, frameshift, nonsense, splice site), including multiexon deletion and duplication[16][17] have been published (HGMD® Professional 2020.1; accessed 15 September 2020).
Because PLAN is caused by biallelic mutations, the disease mechanism is expected to be caused by loss of function. PLA2G6 encodes several isoforms of VIA calcium-independent phospholipase A2, which hydrolyzes the sn-2 ester bond in membrane phospholipids, releasing free fatty acids and 2-lysophospholipids[18] (Figure 1). It is involved in transduction and maintenance of phospholipid homeostasis, releasing docohexaenoic acid (DHA) and arachidonic acid[19]. In addition, roles related to inflammation and immune responses, chemotaxis, vascular relaxation, secretion and apoptosis have been described for this phospholipase[20][21].
3.2. Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN)
Mutations in the C19ORF12 (chromosome 19 open reading frame 12) gene cause MPAN, which is characterized by cognitive decline progressing to dementia, speech and gait disturbances, parkinsonism, optic atrophy and motor axonal neuropathy[22]. The first C19ORF12 mutation discovered was a homozygous 11 bp deletion leading to a truncated protein (c.204_214del11, p.Gly69Argfs*10)) in 13 families from Eastern Europe[23]. Nowadays, about 50 C19ORF12 variants have been found to relate to the MPAN phenotype, including missense/nonsense, indels and splicing mutations (HGMD® Professional 2020.1; accessed 7 October 2020). Although MPAN is considered an AR condition, a single C19ORF12 mutation can lead to the same clinical phenotype as biallelic variants[24]. C19ORF12 synthesizes a small transmembrane protein (17 kDa), whose function remains unclear (Figure 1). It is widely expressed in the brain and adipocytes and localized in the lumen and MAMs (mitochondria associated membranes) of mitochondria, and in the ER.
3.3. Fatty Acid Hydroxylase-Associated Neurodegeneration (FAHN)
Mutations in FA2H (fatty acid 2-hydroxylase) cause AR FAHN. Clinically, FAHN is characterized by ataxia, dystonia, spasticity, ocular abnormalities, cerebellar atrophy, and iron deposition, predominantly in the GP. Cognitive impairment and seizures may be features of the disease[25]. White matter abnormalities, a thinner corpus callosum and supratentorial atrophy seem to be hallmarks shared by the vast majority of the patients[26]. Up to 65 mutations (missense/nonsense, frameshift, splicing and indels) in FA2H have been described (HGMD® Professional 2020.1; accessed 7 October 2020). FA2H encodes a NADPH-dependent mono-oxygenase that colocalizes with the ER membrane. FA2H is crucial during the early stages of brain development. Due to its 2-hydroxylase activity, FA2H produces 2-hydroxylated ceramides and therefore, participates in myelin formation[27] (Figure 1).
4. Autophagosome/Lysosome Regulation
4.1. β-propeller-Associated Neurodegeneration (BPAN)
BPAN disorder is caused by mutations in the X chromosome gene WDR45, which is transmitted in an AD manner. To date, all affected individuals are sporadic cases with no family history (de novo mutations). Clinical features do not always follow the typical pattern for an X-linked disorder. Apparently, affected men, who are carriers of hemizygous WDR45 mutations, are predicted to harbor post-zygotic mutations, suggesting that male patients could be somatic mosaicisms[28]. Affected women may carry either germline or somatic mutations, showing different phenotypic manifestations, probably associated with skewing of X chromosome inactivation[29]. So far, about 112 disease-causing mutations are known in WDR45 (HGMD® Professional 2020.1; accessed 13 October 2020).
Clinically, BPAN is well characterized as a two-stage disease progression. The first stage comprises a global developmental delay in childhood with intellectual disability. Common early comorbidities comprise seizures, spasticity, and epilepsy. The second stage affects all patients in early adulthood, and manifests with progressive dystonia, dementia and parkinsonism characterized by bradykinesia and rigidity without tremor. Brain magnetic resonance imaging (MRI) shows iron accumulation in the substantia nigra and globus pallidus in the early phase. A T1-weighted hyperintense “halo” signal with a central band of hypointensity in the substantia nigra seems to be a specific finding in BPAN. Cerebral atrophy is also reported in most patients. Additional symptoms are sleep disturbance, ocular features and Rett-like hand stereotypies[29].
The WDR45 protein is a member of the WD40 repeat protein family with a β-propeller platform structure. WD40 proteins play a role in coordinating protein–protein interactions in order to perform a variety of functions, such as signal transduction, autophagy or transcriptional regulation. In particular, the WDR45 protein, by binding to phosphatidylinositol-3-phosphate (PtdIns3P), regulates autophagosome formation[30].
4.2. Kufor–Rakeb Syndrome
Kufor–Rakeb disease (KRD) is a very rare early-onset atypical parkinsonism caused by mutations in the ATP13A2 gene with AR inheritance. Symptoms appear before 20 years of age and are characterized by motor symptoms (pyramidal degeneration) and non-motor symptoms (dementia, learning difficulties and hallucinations)[31][32]. About 50 mutations associated with KRD, including additional phenotypes such as neuronal ceroid-lipofuscinosis and hereditary mutations, have been reported in ATP13A2 (HGMD® Professional 2020.1; accessed 16 September 2020)[33]. Most patients with AR parkinsonism do not accumulate iron in the brain despite being clinically symptomatic. Nevertheless, some cases with homozygous mutations in ATP13A2 and evidence of iron deposition in the basal ganglia have been reported, which makes it possible to include KRD in the NBIA group of disorders[34][35]. ATP13A2 encodes for a lysosomal 5P-type ATPase, and is mostly localized in endosomes, lysosomes, and partially, in autophagosomes (Figure 1). Several functions have been attributed to ATP13A2 including homeostasis of manganese, zinc and iron, allowing active transportation across endosomal and lysosomal membranes, mitochondrial bioenergetics and the autophagy-lysosomal pathway. Defects in ATP13A2 may impair the endo-lysosomal and autophagy flux, resulting in the accumulation of insoluble proteins and damaged mitochondria, leading to apoptosis and neuroinflammation.
5. NBIA Forms Caused by Mutations in Iron-Related Genes
5.1. Aceruloplasminemia
Aceruloplasminemia is a rare AR disorder caused by mutations in the CP gene, which encodes for ceruloplasmin (Cp), a multicopper ferroxidase functioning as an iron exporter from cells[36]. Intracellular Fe2+ is transported by ferroportin to transferrin via the ferroxidase activity of ceruloplasmin (Fe2+ -> Fe3+) (Figure 1). Close to 70 mutations (missense, frameshift, splicing, nonsense) have been described in CP (HGMD® Professional 2020.1; accessed 16 September 2020). Homozygous mutations are identified in the vast majority of patients, although compound heterozygosity may be also detected. Even though inheritance is AR, heterozygous carriers may present with a milder clinical picture[37]. Cp function is essential in astrocytes as it is the only ferroxidase in this cell type. Its absence causes remarkable morphological abnormalities, oxidative stress due to iron accumulation, and lipid peroxidation. These features are also present in other brain tissues, being severe in the basal ganglia, thalamus and cerebellum of the patients, which supports toxicity of iron excess[38]. The CP-associated phenotype is characterized by dementia, ataxia, chorea and parkinsonism in adulthood[39]. Microcytic anemia, liver disease and retinopathy are also common clinical features.
5.2. Neuropherritinopathy (NF)
Mutations in the ferritin light chain (FTL) gene cause neuroferritinopathy, an AD inherited disease with adulthood onset. So far, ten nucleotide duplications in exon 4 (HGMD® Professional 2020.1; accessed 10 September 2020), involving COOH-terminal residues of the protein, are associated with NF, and they may account for dystonia, dysarthria, cerebellar signs, parkinsonism, chorea and psychiatric features[40]. Ferritin removes ferrous iron from labile iron pools in cells in order to prevent cellular damage, oxygen reactive species formation, lipid peroxidation, protein aggregation and iron overload, common features of NF (Figure 1). Iron is stored in the form of ferric iron oxide inside ferritin, which is responsible for its release when it is required by cellular pathways. Native human ferritin is a heteropolymer composed of two subunits: FTH1 and FTL. Each cell/organ presents different ratios of these subunits depending on their storage rate and optimization. COOH-terminus modifications seem to result in an alteration of the heteropolymeric structure that prevents iron from binding, and promotes the generation of ferritin aggregates and the formation of inclusion bodies[41]. As this is a consequence of long-term stress conditions, development of the disease is gradual in the patients.
6. Another NBIA Subtype: Woodhouse-Sakati Syndrome
Woodhouse-Sakati syndrome is a rare AR disorder caused by mutations in the DCAF17 gene. To date, a total of 17 variants related to this syndrome have been described, including five missense mutations, six splicing mutations, and 12 small insertions or deletions (HGMD® Professional 2020.1; accessed 8 October 2020). The complex clinical picture is characterized by the effects on SNC, but also on the SNP and the neuroendocrine system. Some of the Woodhouse-Sakati traits are hypogonadism, diabetes mellitus, mental retardation, deafness, alopecia, polyneuropathy and extrapyramidal impairment[42][43]. This gene encodes a nucleolar protein (Figure 1) expressed in many tissues, including the brain, liver, skin and gonads. The function of Dcaf17 is still unknown, but it is related to the Dcaf gene family and is involved in apoptosis, DNA methylation and cell cycle regulation[44].
7. Conclusions
NBIA syndromes share the distinctive feature of iron overload in the brain and present a wide genetic heterogeneity with genes related to disparate pathways. How these different genes can cause the abnormal deposits of iron in the brain is a matter of investigation. However, as usually occurs in many other neurodegenerative disorders, mitochondrial dysfunction may play a vital role in the underlying pathomechanism because of the high energy demand within the brain. NBIA genes directly related to mitochondrial functions are PANK2, COASY and probably, C19ORF12, which ultimately together with PLA2G6 and FA2H are involved in phospholipid membrane synthesis (Figure 1). WDR45 is crucial in the formation/degradation of autophagosomes as well as ATP13A2, a lysosomal protein. Lysosomal activity is essential for iron homeostasis in which FTL and CP take part. As a whole, the disease process in at least seven NBIA forms involves mitochondrial dysfunction, which produces oxidative stress and leads to neuroinflammation. Nonetheless, the list of NBIA genes is growing and the connection between all the players involved in NBIA disorders is unclear. Further studies are necessary with the aim of characterizing targets for therapeutic interventions.
References
- Carmen Espinós; Máximo Ibo Galindo; María Adelaida García-Gimeno; José Santiago Ibáñez-Cabellos; Dolores Martínez-Rubio; José María Millán; Regina Rodrigo; Pascual Sanz; Marta Seco-Cervera; Teresa Sevilla; et al.Andrea TapiaFederico V. Pallardó Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313, 10.3390/antiox9040313.
- Monique A. Johnson; Yien Ming Kuo; Shawn K. Westaway; Susan M. Parker; Katherine H. L. Ching; Jane Gitschier; Susan J. Hayflick; Mitochondrial Localization of Human PANK2 and Hypotheses of Secondary Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration. Annals of the New York Academy of Sciences 2004, 1012, 282-298, 10.1196/annals.1306.023.
- Konstanze Hörtnagel; Holger Prokisch; Thomas Meitinger; An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Human Molecular Genetics 2003, 12, 321-327, 10.1093/hmg/12.3.321.
- Robert A. Egan; Richard G. Weleber; Penelope Hogarth; Allison Gregory; Jason Coryell; Shawn K. Westaway; Jane Gitschier; Soma Das; Susan J. Hayflick; Neuro-Ophthalmologic and Electroretinographic Findings in Pantothenate Kinase-Associated Neurodegeneration (formerly Hallervorden-Spatz Syndrome). American Journal of Ophthalmology 2005, 140, 267.e1-267.e9, 10.1016/j.ajo.2005.03.024.
- Susan J. Hayflick; Neurodegeneration With Brain Iron Accumulation: From Genes to Pathogenesis. Seminars in Pediatric Neurology 2006, 13, 182-185, 10.1016/j.spen.2006.08.007.
- Rafael Fermin Delgado; Pedro Roa Sanchez; Herwin Speckter; Eddy Perez-Then; Ramney Jimenez; Jairo Oviedo; Paulo Roberto Dellani; Bernd Foerster; Peter Stoeter; Missense PANK2 mutation without “Eye of the tiger” sign: MR findings in a large group of patients with pantothenate kinase-associated neurodegeneration (PKAN). Journal of Magnetic Resonance Imaging 2011, 35, 788-794, 10.1002/jmri.22884.
- James Stankiewicz; S. Scott Panter; Mohit Neema; Ashish Arora; Courtney E. Batt; Rohit Bakshi; Iron in chronic brain disorders: Imaging and neurotherapeutic implications. Neurotherapeutics 2007, 4, 371-386, 10.1016/j.nurt.2007.05.006.
- Alessandro Malandrini; Gian Maria Fabrizi; Paola Bartalucci; Claudio Salvadori; Gianna Berti; Gian Carlo Guazzi; Clinicopathological study of familial late infantile Hallervorden-Spatz disease: a particular form of neuroacanthocytosis. Child's Nervous System 1996, 12, 155-160, 10.1007/bf00266820.
- Michael C. Kruer; Mark Hiken; Allison Gregory; Alessandro Malandrini; David Clark; Penny Hogarth; Marjorie Grafe; Susan J. Hayflick; Randall L. Woltjer; Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011, 134, 947-958, 10.1093/brain/awr042.
- Sabrina Dusi; Lorella Valletta; Tobias B. Haack; Yugo Tsuchiya; Paola Venco; Sebastiano Pasqualato; Paola Goffrini; Marco Tigano; Nikita Demchenko; Thomas Wieland; et al.Thomas SchwarzmayrTim M. StromFederica InvernizziBarbara GaravagliaAllison GregoryLynn SanfordJeffrey HamadaConceicao BettencourtHenry HouldenLuisa ChiappariniGiovanna ZorziManju A. KurianNardo NardocciHolger ProkischSusan HayflickIvan GoutValeria Tiranti Exome Sequence Reveals Mutations in CoA Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation. The American Journal of Human Genetics 2014, 94, 11-22, 10.1016/j.ajhg.2013.11.008.
- Christina Evers; Angelika Seitz; Birgit Assmann; Thomas Opladen; Stephanie Karch; Katrin Hinderhofer; Martin Granzow; Nagarajan Paramasivam; Roland Eils; Nicolle Diessl; et al.Claus R. BartramUte Moog Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger's eye. American Journal of Medical Genetics Part A 2017, 173, 1878-1886, 10.1002/ajmg.a.38252.
- Susan J. Hayflick; Manju A. Kurian; Penelope Hogarth; Neurodegeneration with brain iron accumulation. Handbook of Clinical Neurology 2018, 147, 293-305, 10.1016/b978-0-444-63233-3.00019-1.
- M.A. Illingworth; E. Meyer; W.K. Chong; A.Y. Manzur; L.J. Carr; R. Younis; C. Hardy; F. McDonald; A.M. Childs; B. Stewart; et al.D. WarrenR. KneenM.D. KingS.J. HayflickM.A. Kurian PLA2G6-associated neurodegeneration (PLAN): Further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Molecular Genetics and Metabolism 2014, 112, 183-189, 10.1016/j.ymgme.2014.03.008.
- Mario Mascalchi; Francesco Mari; Beatrice Berti; Emanuele Bartolini; Matteo Lenge; Andrea Bianchi; Laura Antonucci; Filippo M. Santorelli; Barbara Garavaglia; Renzo Guerrini; et al. Fast Progression of Cerebellar Atrophy in PLA2G6-Associated Infantile Neuronal Axonal Dystrophy. The Cerebellum 2017, 16, 742-745, 10.1007/s12311-017-0843-z.
- A Mubaidin; E Roberts; Daniel Hampshire; M Dehyyat; A Shurbaji; M Mubaidien; A Jamil; A Al-Din; A Kurdi; C G Woods; et al. Karak syndrome: a novel degenerative disorder of the basal ganglia and cerebellum. Journal of Medical Genetics 2003, 40, 543-546, 10.1136/jmg.40.7.543.
- Danielle Crompton; Pauline K. Rehal; Lesley MacPherson; Katharine Foster; Peter Lunt; Imelda Hughes; Angela F. Brady; Michael G. Pike; Susanna De Gressi; Neil V. Morgan; et al. Multiplex ligation-dependent probe amplification (MLPA) analysis is an effective tool for the detection of novel intragenic PLA2G6 mutations: Implications for molecular diagnosis. Molecular Genetics and Metabolism 2010, 100, 207-212, 10.1016/j.ymgme.2010.02.009.
- Toshiyuki Yamamoto; Keiko Shimojima; Takashi Shibata; Mari Akiyama; Makio Oka; Tomoyuki Akiyama; Harumi Yoshinaga; Katsuhiro Kobayashi; Novel PLA2G6 mutations associated with an exonic deletion due to non-allelic homologous recombination in a patient with infantile neuroaxonal dystrophy. Human Genome Variation 2015, 2, 15048, 10.1038/hgv.2015.48.
- Konstantin R. Malley; Olga Koroleva; Ian Miller; Ruslan Sanishvili; Christopher M. Jenkins; Richard W. Gross; Sergey Korolev; The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nature Communications 2018, 9, 1-11, 10.1038/s41467-018-03193-0.
- Jin Tang; Ronald W. Kriz; Neil Wolfman; Mary Shaffer; Jasbir Seehra; Simon S. Jones; A Novel Cytosolic Calcium-independent Phospholipase A2 Contains Eight Ankyrin Motifs. Journal of Biological Chemistry 1997, 272, 8567-8575, 10.1074/jbc.272.13.8567.
- Sasanka Ramanadham; Tomader Ali; Jason Ashley; Robert Bone; William D. Hancock; Xiaoyong Lei; Calcium-independent phospholipases A2 and their roles in biological processes and diseases. Journal of Lipid Research 2015, 56, 1643-1668, 10.1194/jlr.r058701.
- John Turk; Tayleur D. White; Alexander J. Nelson; Xiaoyong Lei; Sasanka Ramanadham; iPLA2β and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2018, 1864, 846-860, 10.1016/j.bbalip.2018.10.010.
- Monika Hartig; Holger Prokisch; Thomas Meitinger; Thomas Klopstock; Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN). International Review of Neurobiology 2013, 110, 73-84, 10.1016/b978-0-12-410502-7.00004-1.
- Monika B. Hartig; Arcangela Iuso; Tobias Haack; Tomasz Kmiec; Elzbieta Jurkiewicz; Katharina Heim; Sigrun Roeber; Victoria Tarabin; Sabrina Dusi; Malgorzata Krajewska-Walasek; et al.Sergiusz JozwiakMaja HempelJuliane WinkelmannMatthias ElstnerKonrad OexleThomas KlopstockWolfgang Mueller-FelberThomas GasserClaudia TrenkwalderValeria TirantiHans KretzschmarGerd SchmitzTim M. StromThomas MeitingerHolger Prokisch Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation. The American Journal of Human Genetics 2011, 89, 543-550, 10.1016/j.ajhg.2011.09.007.
- Allison Gregory; Mitesh Lotia; Suh Young Jeong; Rachel Fox; Dolly Zhen; Lynn Sanford; Jeff Hamada; Amir Jahic; Christian Beetz; Alison Freed; et al.Manju A. KurianThomas CullupMarlous C. M. Van Der WeijdenVy NguyenNaly SetthavongsackDaphne GarciaVictoria KrajbichThao PhamRandy WoltjerBenjamin P. GeorgeKelly Q. MinksAlexander R. PaciorkowskiPenelope HogarthJoseph JankovicSusan J. Hayflick Autosomal dominant mitochondrial membrane protein‐associated neurodegeneration (MPAN). Molecular Genetics & Genomic Medicine 2019, 7, e00736, 10.1002/mgg3.736.
- Michael C. Kruer; Coro Paisán-Ruiz; Nathalie Boddaert; Moon Y. Yoon; Hiroko Hama; Allison Gregory Ms; Alessandro Malandrini; Randall L. Woltjer Md; Arnold Munnich; Stephanie Gobin; et al.Brenda J. PolsterSilvia PalmeriSimon EdvardsonJohn HardyHenry HouldenSusan J. Hayflick Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Annals of Neurology 2010, 68, 611-618, 10.1002/ana.22122.
- Tim W. Rattay; Tobias Lindig; Jonathan Baets; Katrien Smets; Tine Deconinck; Anne S Söhn; Konstanze Hörtnagel; Kathrin N Eckstein; Sarah Wiethoff; Jennifer Reichbauer; et al.Marion Döbler-NeumannIngeborg Krägeloh-MannMichaela Auer-GrumbachBarbara PleckoAlexander MünchauBernd WilkenMarc JanauschekAnne-Katrin GieseJan L De BleeckerEls OrtibusMartine DebyserAdolfo Lopez De MunainAurora PujolMaria Teresa BassiMaria Grazia D’AngeloPeter De JongheStephan ZüchnerPeter BauerLudger SchölsRebecca Schüle FAHN/SPG35: a narrow phenotypic spectrum across disease classifications. Brain 2019, 142, 1561-1572, 10.1093/brain/awz102.
- Matthias Eckhardt; Afshin Yaghootfam; Simon Ngamli Fewou; Inge Zöller; Volkmar Gieselmann; A mammalian fatty acid hydroxylase responsible for the formation of α-hydroxylated galactosylceramide in myelin. Biochemical Journal 2005, 388, 245-254, 10.1042/bj20041451.
- Tobias B. Haack; Penelope Hogarth; Michael C. Kruer; Allison Gregory; Thomas Wieland; Thomas Schwarzmayr; Elisabeth Graf; Lynn Sanford; Esther Meyer; Eleanna Kara; et al.Stephan M. CunoSami I. HarikVasuki H. DanduNardo NardocciGiovanna ZorziTodd DunawayMark TarnopolskySteven SkinnerSteven FruchtEra HanspalConnie Schrander-StumpelDelphine HéronCyril MignotBarbara GaravagliaKailash BhatiaJohn HardyTim M. StromNathalie BoddaertHenry H. HouldenManju A. KurianThomas MeitingerHolger ProkischSusan J. Hayflick Exome Sequencing Reveals De Novo WDR45 Mutations Causing a Phenotypically Distinct, X-Linked Dominant Form of NBIA. The American Journal of Human Genetics 2012, 91, 1144-1149, 10.1016/j.ajhg.2012.10.019.
- Susan J. Hayflick; Michael C. Kruer; Allison Gregory; Tobias B. Haack; Manju A. Kurian; Henry H. Houlden; James Anderson; Nathalie Boddaert; Lynn Sanford; Sami I. Harik; et al.Vasuki H. DanduNardo NardocciGiovanna ZorziTodd DunawayMark TarnopolskySteven SkinnerKenton R. HoldenSteven FruchtEra HanspalConnie Schrander-StumpelCyril MignotDelphine HéronDawn E. SaundersMargaret KaminskaJean-Pierre LinKarine LascellesStephan M. CunoEsther MeyerBarbara GaravagliaKailash BhatiaRajith De SilvaSarah CrispPeter LuntMartyn CareyJohn HardyThomas MeitingerHolger ProkischPenelope Hogarth Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013, 136, 1708-1717, 10.1093/brain/awt095.
- Shintaro Maeda; Chinatsu Otomo; Takanori Otomo; The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 2019, 8, 555441, 10.1101/555441.
- A S Najim Al-Din; A. Wriekat; A. Mubaidin; M. Dasouki; M. Hiari; Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurologica Scandinavica 2009, 89, 347-352, 10.1111/j.1600-0404.1994.tb02645.x.
- Alfredo Ramirez; André Heimbach; Jan Gründemann; Barbara Stiller; Dan Hampshire; L Pablo Cid; Ingrid Goebel; Ammar F Mubaidin; Abdul-Latif Wriekat; Jochen Roeper; et al.Amir Al-DinAxel HillmerMeliha KarsakBirgit LissC Geoffrey WoodsMaria I BehrensChristian Kubisch Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature Genetics 2006, 38, 1184-1191, 10.1038/ng1884.
- Jose Bras; Alain Verloes; Susanne A. Schneider; Sara E. Mole; Rita J. Guerreiro; Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Human Molecular Genetics 2012, 21, 2646-2650, 10.1093/hmg/dds089.
- Maria I. Behrens; Norbert Brüggemann; Pedro Chana; Pablo Venegas; Marianne Kägi; Teresa Parrao; Patricia Orellana; Cristian Garrido; Cecilia V. Rojas; Jan Hauke; et al.Eric HahnenRafael GonzálezNicolas SelemeVerónica FernándezAlexander SchmidtFerdinand BinkofskiDetlef KömpfChristian KubischJohann HagenahChristine KleinAlfredo Ramirez Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Movement Disorders 2010, 25, 1929-1937, 10.1002/mds.22996.
- Susanne A. Schneider Md; Coro Paisan-Ruiz; Niall P. Quinn; Andrew J. Lees; Henry Houlden; John Hardy; Kailash P. Bhatia; ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Movement Disorders 2010, 25, 979-984, 10.1002/mds.22947.
- Sonia Elevi; Dario Efinazzi; Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Frontiers in Pharmacology 2014, 5, 99, 10.3389/fphar.2014.00099.
- Alisdair McNeill; Massimo Pandolfo; Jens Kuhn; Huifang Shang; Hiroaki Miyajima; The Neurological Presentation of Ceruloplasmin Gene Mutations. European Neurology 2008, 60, 200-205, 10.1159/000148691.
- Satoshi Kono; Hiroaki Miyajima; Molecular and pathological basis of aceruloplasminemia. Biological Research 2006, 39, 15-23, 10.4067/s0716-97602006000100003.
- Luis F. Gonzalez-Cuyar; George Perry; Hiroaki Miyajima; Craig S. Atwood; Marcela Riveros-Angel; Patrick F. Lyons; Sandra L. Siedlak; Mark A. Smith; Rudy J. Castellani; Redox active iron accumulation in aceruloplasminemia. Neuropathology 2008, 28, 466-471, 10.1111/j.1440-1789.2008.00901.x.
- Barry B. Muhoberac; Ruben Vidal; Iron, Ferritin, Hereditary Ferritinopathy, and Neurodegeneration. Frontiers in Neuroscience 2019, 13, 1195, 10.3389/fnins.2019.01195.
- Barry B. Muhoberac; Ruben Evidal; Abnormal iron homeostasis and neurodegeneration. Frontiers in Aging Neuroscience 2013, 5, 32, 10.3389/fnagi.2013.00032.
- Anas M. Alazami; Amr Al-Saif; Abdulaziz Al-Semari; Saeed Bohlega; Soumaya Zlitni; Fatema Alzahrani; Prashant Bavi; Namik Kaya; Dilek Colak; Hanif Khalak; et al.Andy BaltusBorut PeterlinSumita DandaKailash P. BhatiaSusanne A. SchneiderNadia SakatiChristopher A. WalshFutwan Al-MohannaBrian MeyerFowzan S. Alkuraya Mutations in C2orf37, Encoding a Nucleolar Protein, Cause Hypogonadism, Alopecia, Diabetes Mellitus, Mental Retardation, and Extrapyramidal Syndrome. The American Journal of Human Genetics 2008, 83, 684-691, 10.1016/j.ajhg.2008.10.018.
- F. Gurbuz; S. Desai; F. Diao; D. Turkkahraman; F. Wranitz; Michelle Wood-Trageser; Y.-H. Shin; L. Damla Kotan; H. Jiang; S. Witchel; et al.N. GurtuncaS. YatsenkoD. MysliwecK. TopalogluA. Rajkovic Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clinical Genetics 2017, 93, 853-859, 10.1111/cge.13183.
- Jennifer Lee; Pengbo Zhou; DCAFs, the Missing Link of the CUL4-DDB1 Ubiquitin Ligase. Molecular Cell 2007, 26, 775-780, 10.1016/j.molcel.2007.06.001.

