 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Min Wang | -- | 416 | 2022-07-06 02:41:23 | | | |
| 2 | Min Wang | + 1738 word(s) | 2154 | 2022-07-06 03:32:42 | | | | |
| 3 | Beatrix Zheng | -6 word(s) | 2148 | 2022-07-06 04:30:57 | | | | |
| 4 | Beatrix Zheng | + 2 word(s) | 2150 | 2022-07-06 04:32:22 | | |
Video Upload Options
Urolithin A (Uro A) is a dietary metabolite of the intestinal microbiota following the ingestion of plant-based food ingredients ellagitannins and ellagic acid in mammals. Accumulating studies have reported its multiple potential health benefits in a broad range of diseases, including cardiovascular disease, cancer, cognitive impairment, and diabetes.
1. Reduces the Expression of Pancreatic Inflammatory Factors
| Disease Model | Treatment | Metabolic Response | Ref. |
|---|---|---|---|
| Spontaneous CP in male Wistar Bonn/Kobori rats | 100 mg/kg BW/day orally administered with EA for 10 weeks |
|
[3] |
| PSCs were isolated from rat pancreas tissue to culture activated, myofibroblast-like phenotype | 1–25 μg/mL EA |
|
[4] |
| L-arginine induced AP in rats | 85 mg/kg orally administered with EA |
|
[5] |
| MIN6 β-cell inflammations were induced using 25 mM glucose and 0.5 mM palmitic acid | Uro A |
|
[6] |
| Alcohol-associated CP in C56BL6/J mice | Administered during the last 3 weeks of alcohol-associated CP induction |
|
[7] |
| DM in male C57BL/6 mice was achieved by a HFD and intraperitoneal STZ injections | 50 mg/kg BW/day orally administered with Uro A for 8 weeks |
|
[8] |
| Human PDAC cell lines; PDAC mice were achieved by injecting PANC1 cells into the flank of 6-week-old Fox1-nu/nu mice | 0–100 μM; 20 mg/kg BW/day (5 days/week) orally administered with Uro A |
|
[9] |
| PKT (Ptf1acre/+; LSL-KrasG12D; Tgfbr2fl/fl) mice, an aggressive genetically engineered PDAC mouse model | Orally administered with Uro A for 5 weeks |
|
[10] |
| Neonatal STZ-induced non-obese T2DM rats | 25–100 mg/kg BW orally administered with EA |
|
[11] |
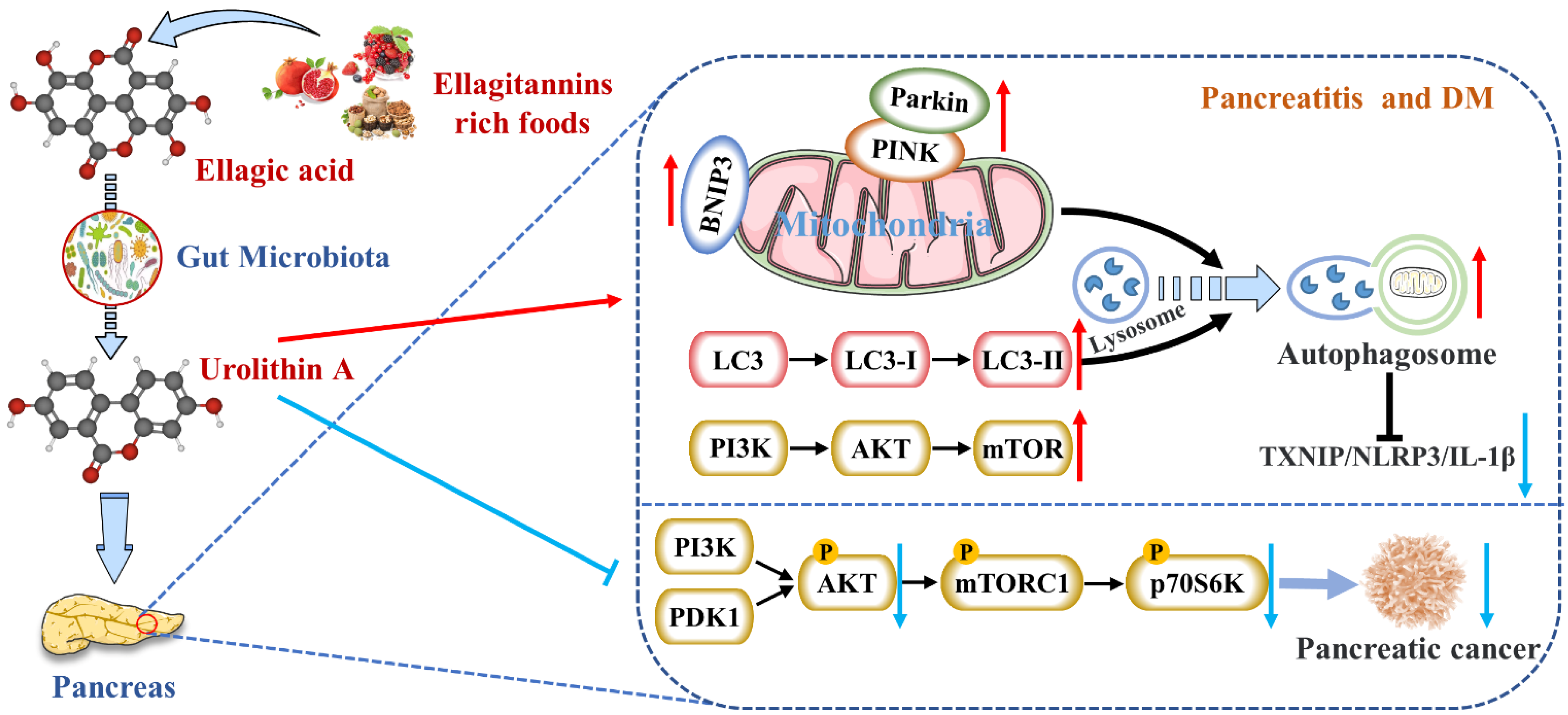
Nevertheless, studies on reducing pancreatic inflammation by Uro A had only been verified in animals and cells without clinical studies. The upstream mediators of Uro A’s anti-inflammatory effects, including the NF-κB and AhR-Nrf2 pathways, were mainly studied in vitro [16]. Nevertheless, the mechanisms of Uro A action in the context of inflammation seemed to vary with tissues and conditions. Hence, the differences in Uro A’s mitigation degree and mechanism on AP and CP need to be further explored.
2. Activates Autophagy and Maintains Mitochondrial Function in the Pancreas
3. Inhibits Endoplasmic Reticulum Stress in the Pancreas
4. Inhibits the Occurrence and Development of Pancreatic Tumors
5. Protects Pancreatic β Cells
References
- Sandovici, I.; Hammerle, C.M.; Cooper, W.N.; Smith, N.H.; Tarry-Adkins, J.L.; Dunmore, B.J.; Bauer, J.; Andrews, S.R.; Yeo, G.S.; Ozanne, S.E.; et al. Ageing is associated with molecular signatures of inflammation and type 2 diabetes in rat pancreatic islets. Diabetologia 2016, 59, 502–511.
- Janjuha, S.; Singh, S.P.; Tsakmaki, A.; Gharavy, S.N.M.; Murawala, P.; Konantz, J.; Birke, S.; Hodson, D.J.; Rutter, G.A.; Bewick, G.A.; et al. Age-related islet inflammation marks the proliferative decline of pancreatic beta-cells in zebrafish. Elife 2018, 7, e32965.
- Suzuki, N.; Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Shimosegawa, T. Ellagic Acid Inhibits Pancreatic Fibrosis in Male Wistar Bonn/Kobori Rats. Dig. Dis. Sci. 2009, 54, 802–810.
- Masamune, A.; Satoh, M.; Kikuta, K.; Suzuki, N.; Satoh, K.; Shimosegawa, T. Ellagic acid blocks activation of pancreatic stellate cells. Biochem. Pharmacol. 2005, 70, 869–878.
- Yilmaz, E.E.; Bozdag, Z.; Ibiloglu, I.; Arikanoglu, Z.; Yazgan, U.C.; Kaplan, I.; Gumus, M.; Atamanalp, S.S. Therapeutic effects of ellagic acid on L-arginin induced acute pancreatitis. Acta Cir. Bras. 2016, 31, 396–401.
- Zhang, Y.; Aisker, G.; Dong, H.; Halemahebai, G.; Zhang, Y.; Tian, L. Urolithin A suppresses glucolipotoxicity-induced ER stress and TXNIP/NLRP3/IL-1beta inflammation signal in pancreatic beta cells by regulating AMPK and autophagy. Phytomedicine 2021, 93, 153741.
- Mehra, S.; Srinivasan, S.; Dai, X.; Dosch, A.; Singh, S.; Bianchi, A.; Silva, I.D.C.; Datta, J.; VanSaun, M.N.; Merchant, N.; et al. Protective Effects of Urolithin A on Alcoholic Chronic Pancreatitis via Inhibiting PI3K/AKT/mTOR Signaling Pathway. Pancreas 2021, 50, 1082.
- Tuohetaerbaike, B.; Zhang, Y.; Tian, Y.; Zhang, N.N.; Kang, J.; Mao, X.; Zhang, Y.; Li, X. Pancreas protective effects of Urolithin A on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/mTOR signaling pathway. J. Ethnopharmacol. 2020, 250, 112479.
- Totiger, T.M.; Srinivasan, S.; Jala, V.R.; Lamichhane, P.; Dosch, A.R.; Gaidarski, A.A., III; Joshi, C.; Rangappa, S.; Castellanos, J.; Vemula, P.K.; et al. Urolithin A, a Novel Natural Compound to Target PI3K/AKT/mTOR Pathway in Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 301–311.
- Srinivasan, S.; Jala, V.; Honnenahally, K.; Castellanos, J.; Vermula, P.K.; VanSaun, M.; Merchant, N.; Nagathihalli, N. Urolithin A prevents pancreatic tumor growth and increases survival by inhibiting PI3K/PDK1 and STAT3 signaling. Cancer Res. 2017, 77, 5259.
- Fatima, N.; Hafizur, R.M.; Hameed, A.; Ahmed, S.; Nisar, M.; Kabir, N. Ellagic acid in Emblica officinalis exerts anti-diabetic activity through the action on beta-cells of pancreas. Eur. J. Nutr. 2017, 56, 591–601.
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem. Toxicol. 2017, 108, 289–297.
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699.
- Larrosa, M.; González-Sarrías, A.; Yáñez-Gascón, M.J.; Selma, M.V.; Azorín-Ortuño, M.; Toti, S.; Tomas-Barberan, F.; Dolara, P.; Espín, J.C. Anti-inflammatory properties of a pomegranate extract and its metabolite urolithin-A in a colitis rat model and the effect of colon inflammation on phenolic metabolism. J. Nutr. Biochem. 2010, 21, 717–725.
- Wu, D.; Yan, Z.B.; Cheng, Y.G.; Zhong, M.W.; Liu, S.Z.; Zhang, G.Y.; Hu, S.Y. Deactivation of the NLRP3 inflammasome in infiltrating macrophages by duodenal-jejunal bypass surgery mediates improvement of beta cell function in type 2 diabetes. Metabolism 2018, 81, 1–12.
- Singh, R.; Chandrashekharappa, S.; Bodduluri, H.; Baby, B.V.; Hegde, B.; Kotla, N.G.; Hiwale, A.A.; Saiyed, T.; Patel, P.; Vijay-Kumar, M.; et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun. 2019, 10, 89.
- Vezza, T.; Díaz-Pozo, P.; Canet, F.; de Marañón, A.M.; Abad-Jiménez, Z.; García-Gargallo, C.; Roldan, I.; Solá, E.; Bañuls, C.; López-Domènech, S.; et al. The Role of Mitochondrial Dynamic Dysfunction in Age-Associated Type 2 Diabetes. World J. Men’s Health 2022, 40.
- De Tata, V. Age-related impairment of pancreatic Beta-cell function: Pathophysiological and cellular mechanisms. Front. Endocrinol. 2014, 5, 138.
- Abuarab, N.; Munsey, T.S.; Jiang, L.-H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161.
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects Against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560.
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022.
- Luan, P.; D’Amico, D.; Andreux, P.A.; Laurila, P.-P.; Wohlwend, M.; Li, H.; de Lima, T.I.; Place, N.; Rinsch, C.; Zanou, N.; et al. Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci. Transl. Med. 2021, 13, eabb0319.
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412.
- Zhang, Y.; Zhang, Y.; Halemahebai, G.; Tian, L.; Dong, H.; Aisker, G. Urolithin A: A pomegranate metabolite, protects pancreatic beta cells from apoptosis by activating autophagy. J. Ethnopharmacol. 2021, 272, 113628.
- Marrocco, V.; Tran, T.; Zhu, S.; Choi, S.H.; Gamo, A.M.; Li, S.; Fu, Q.; Cunado, M.D.; Roland, J.; Hull, M.; et al. A small molecule UPR modulator for diabetes identified by high throughput screening. Acta Pharm. Sin. B 2021, 11, 3983–3993.
- Li, J.; Zheng, Y.; Yan, P.; Song, M.; Wang, S.; Sun, L.; Liu, Z.; Ma, S.; Izpisua Belmonte, J.C.; Chan, P.; et al. A single-cell transcriptomic atlas of primate pancreatic islet aging. Natl. Sci. Rev. 2021, 8, 127.
- Mihailidou, C.; Chatzistamou, I.; Papavassiliou, A.G.; Kiaris, H. Modulation of Pancreatic Islets’ Function and Survival During Aging Involves the Differential Regulation of Endoplasmic Reticulum Stress by p21 and CHOP. Antioxid. Redox Signal. 2017, 27, 185–200.
- Veeraraghavan, J.; Natarajan, M.; Lagisetty, P.; Awasthi, V.; Herman, T.S.; Aravindan, N. Impact of Curcumin, Raspberry Extract, and Neem Leaf Extract on Rel Protein-Regulated Cell Death/Radiosensitization in Pancreatic Cancer Cells. Pancreas 2011, 40, 1107–1119.
- Cheng, H.; Lu, C.L.; Tang, R.B.; Pan, Y.M.; Bao, S.H.; Qiu, Y.D.; Xie, M. Ellagic acid inhibits the proliferation of human pancreatic carcinoma PANC-1 cells in vitro and in vivo. Oncotarget 2017, 8, 12301–12310.
- Tomas-Barberan, F.A.; Gonzalez-Sarrias, A.; Garcia-Villalba, R.; Nunez-Sanchez, M.A.; Selma, M.V.; Garcia-Conesa, M.T.; Espin, J.C. Urolithins, the rescue of “old” metabolites to understand a “new” concept, Metabotypes as a nexus among phenolic metabolism, microbiota dysbiosis, and host health status. Mol. Nutr. Food Res. 2017, 61, 1500901.
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031.
- Wong, M.H.; Xue, A.; Baxter, R.C.; Pavlakis, N.; Smith, R.C. Upstream and Downstream Co-inhibition of Mitogen-Activated Protein Kinase and PI3K/Akt/mTOR Pathways in Pancreatic Ductal Adenocarcinoma. Neoplasia 2016, 18, 425–435.
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640.
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80.
- Albasher, G.; Alkahtani, S.; Al-Harbi, L.N. Urolithin A prevents streptozotocin-induced diabetic cardiomyopathy in rats by activating SIRT1. Saudi J. Biol. Sci. 2022, 29, 1210–1220.
- Xiao, Y.; Li, K.; Bian, J.; Liu, H.; Zhai, X.; El-Omar, E.; Han, L.; Gong, L.; Wang, M. Urolithin A Attenuates Diabetes-Associated Cognitive Impairment by Ameliorating Intestinal Barrier Dysfunction via N-glycan Biosynthesis Pathway. Mol. Nutr. Food Res. 2022, 66, 2100863.
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.-J.; et al. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death Differ. 2021, 28, 184–202.
- Xu, Z.; Li, S.; Li, K.; Wang, X.; Li, X.; An, M.; Yu, X.; Long, X.; Zhong, R.; Liu, Q.; et al. Urolithin A ameliorates diabetic retinopathy via activation of the Nrf2/HO-1 pathway. Endocr. J. 2022, EJ21-0490.
- Zhou, J.; Zhang, C.; Zheng, G.-H.; Qiu, Z. Emblic Leafflower (Phyllanthus emblica L.) Fruits Ameliorate Vascular Smooth Muscle Cell Dysfunction in Hyperglycemia: An Underlying Mechanism Involved in Ellagitannin Metabolite Urolithin A. Evid.-Based Complement. Altern. Med. 2018, 2018, 8478943.
- Abdel-Hamid, A.A.M.; El-Firgany, A.E.-D.L. Hydroxychloroquine hindering of diabetic isletopathy carries its signature on the inflammatory cytokines. J. Mol. Histol. 2016, 47, 183–193.
- Tuduri, E.; Soriano, S.; Almagro, L.; Garcia-Heredia, A.; Rafacho, A.; Alonso-Magdalena, P.; Nadal, A.; Quesada, I. The Effects of Aging on Male Mouse Pancreatic beta-Cell Function Involve Multiple Events in the Regulation of Secretion, Influence of Insulin Sensitivity. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 405–415.
- Pan, F.; He, X.; Feng, J.; Cui, W.; Gao, L.; Li, M.; Yang, H.; Wang, C.; Hu, Y. Peptidome analysis reveals the involvement of endogenous peptides in mouse pancreatic dysfunction with aging. J. Cell. Physiol. 2019, 234, 14090–14099.


