 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Whitney Goldsberry | + 9176 word(s) | 9176 | 2019-06-14 11:59:40 | | | |
| 2 | Bruce Ren | -3685 word(s) | 5491 | 2020-10-29 09:22:28 | | | | |
| 3 | Bruce Ren | Meta information modification | 5491 | 2020-10-29 09:23:55 | | |
Video Upload Options
Alterations in the Wnt signaling pathway are associated with the advancement of cancers; however, the exact mechanisms responsible remain largely unknown. It has recently been established that heightened intratumoral Wnt signaling correlates with tumor immunomodulation and immune suppression, which likely contribute to the decreased efficacy of multiple cancer therapeutics. Here, we review available literature pertaining to connections between Wnt pathway activation in the tumor microenvironment and local immunomodulation. We focus specifically on preclinical and clinical data supporting the hypothesis that strategies targeting Wnt signaling could act as adjuncts for cancer therapy, either in combination with chemotherapy or immunotherapy, in a variety of tumor types.
1. Introduction
The Wnt/β-catenin signaling pathway plays a vital role in many cellular functions. Alterations in the pathway are associated with the advancement of cancers. Wnt pathway aberrations have also been correlated with changes in the tumor microenvironment, such as immune evasion, and this has been the topic of two recent reviews [1,2]. The exact mechanisms by which aberrant Wnt signaling modulates anti-tumor immunity need to be further elucidated to facilitate the optimization of cancer therapies. Here, we provide an overview of Wnt signaling pathways and general anti-tumor immunity, then summarize recent literature illustrating how Wnt modulation of the tumor microenvironment (TME) and immune function are being targeted in an attempt to improve cancer treatment outcomes—including both chemotherapies and immunotherapies—for patients with advanced disease. Although there is evidence linking Wnt signaling with metabolic changes that may also impact anti-tumor immunity, we have not addressed this specific topic here, as these findings were recently discussed in detail elsewhere [3,4].
2. The Wnt Pathways
Wnt pathways act as critical signal transduction cascades that modulate embryonic development, adult homeostasis, stem cell control, and wound repair [5]. The first Wnt proteins were discovered in 1982 [6]. There are now 19 known Wnt ligands in mammals, consisting of different glycoproteins, approximately 350–400 amino acids in length, which are highly conserved across many species [7]. The pathways stimulated by these ligands result in gene expression changes that affect the cell’s cytoskeleton and proliferation, while acting as directional growth factors [8]. Due to this important role in cellular regulation, alterations in Wnt signaling consequently may lead to many human diseases, from congenital malformations to nervous system disorders to malignancies, making this pathway a highly desired therapeutic target from a multitude of perspectives. A large portion of Wnt targeted therapies in development focus on Wnt inhibition, while others are Wnt pathway enhancers. A more thorough understanding of the downstream effects of Wnt signaling is necessary for optimal therapeutic manipulation.
As an initial step in this signaling cascade, Wnt ligands must be processed and exported (Figure 1). Wnt proteins are modified by an attachment of a lipid, palmitoleic acid [9]. This modification is performed by the enzyme Porcupine (PORCN), located in the cellular endoplasmic reticulum [9]. This lipid addition is thought to assist in extracellular membrane attraction and act as a binding motif during ligand–receptor interactions [10]. Once the lipid attachment is completed, Wnt protein is then transported to the plasma membrane for secretion [11]. Specific mechanisms of Wnt extracellular transportation are still being investigated, but these may involve secretory vesicle or exosomal incorporation [12].
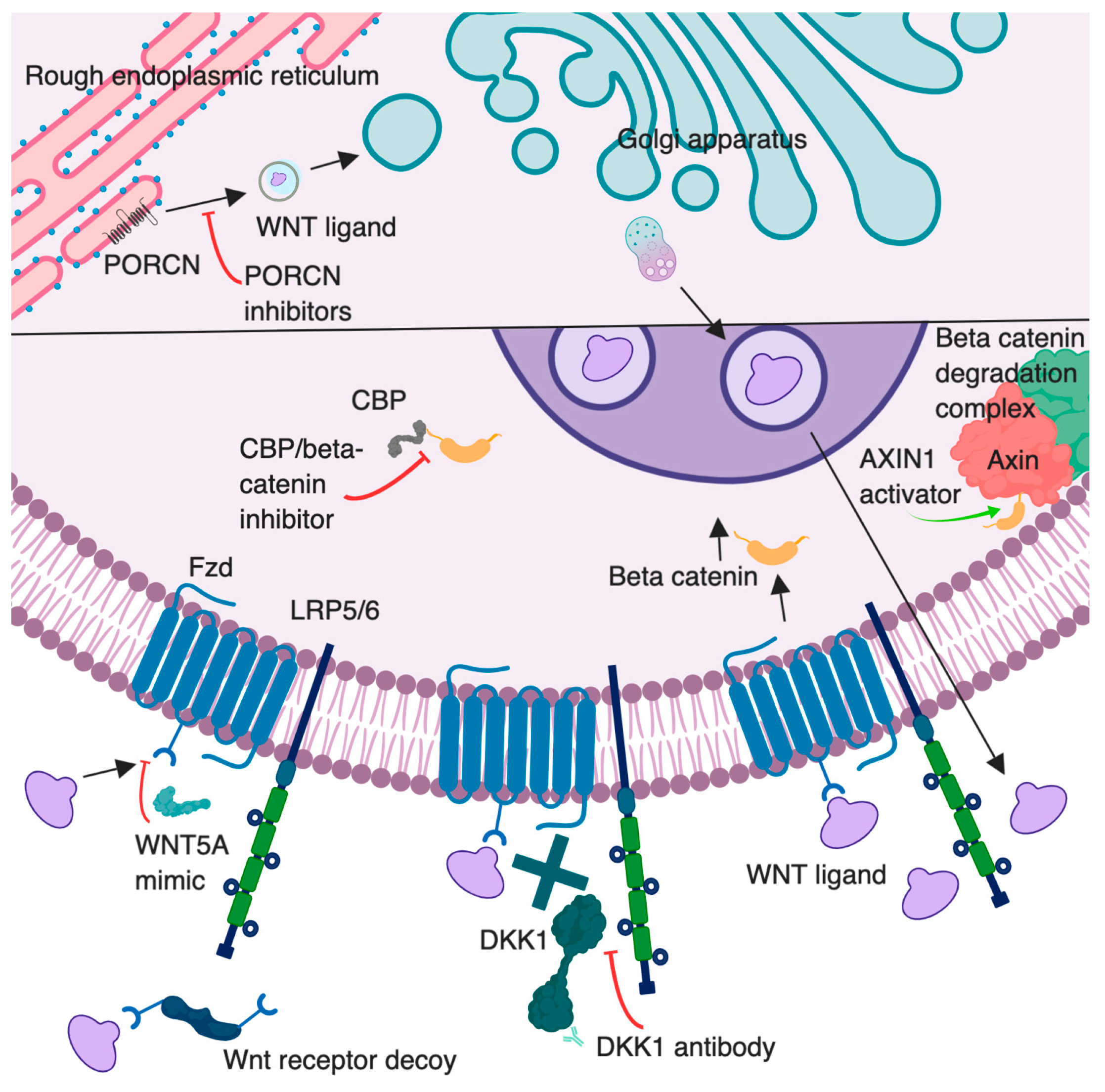
Figure 1. Overview of the canonical Wnt signaling pathway illustrating therapeutic intervention points targeted by Wnt modulators. The lower half of the figure represents a zoomed in portion of the cell. Porcupine (PORCN) inhibitors (e.g., LGK974) block secretion of WNTs by inhibiting their palmitoylation. AXIN1 activators (e.g., niclosamide) promote β-catenin degradation. Dickkopf 1 (DKK1) antibodies may increase Wnt/β-catenin signaling by blocking DKK1 binding to LRP5/6; beneficial effects in cancer may be through inhibition of DKK1 binding to CKAP4 or indirect effects on immune cells. Wnt receptor decoys (e.g., OMP54F28) prevent Wnt binding to Fzd receptors. WNT5A mimic (Foxy-5) is a peptide that activates noncanonical Wnt signals. CBP/beta-catenin inhibitors (e.g., PRI-724) disrupt the interaction between CREB-binding protein (CBP) and β-catenin. Created with BioRender.com. Proteins and cell compartments are not drawn to scale.
2.1. The Canonical Pathway
Traditionally, Wnt pathways are associated with intracellular β-catenin stabilization [13]. Cytoplasmic accumulation of β-catenin allows for its nuclear translocation, resulting in upregulation of Wnt-responsive genes. The signaling cascade begins when Wnt ligands bind to the transmembrane receptor Frizzled (Fzd or Fz) and the coreceptor low-density lipoprotein receptor-related protein-5/6 (LRP5/6) [14] (Figure 1). After this interaction, the cytoplasmic tail of LRP recruits Axin. Disheveled (Dvl), a protein that may bind to Fzd, may act as a platform for this LRP/Axin interaction [5]. Axin is a component of the β-catenin degradation complex. This complex additionally consists of adenomatous polyposis coli (APC), casein kinase 1α (CK1α), and glycogen synthase kinase 3β (GSK3β). When functioning properly, the β-catenin degradation complex phosphorylates the amino-terminal region of β-catenin. This causes recognition of β-catenin by β-Trcp, an E3 ubiquitin ligase subunit, resulting in the ubiquitination and proteasomal degradation of β-catenin [15,16]. Once the complex is inactive, a stable accumulation of β-catenin occurs in the cell cytosol, culminating in β-catenin translocation into the cell nucleus [17]. Here, β-catenin can bind to the transcription factor T cell factor/lymphoid enhancer-binding factor (TCF/Lef1), activating the transcription of Wnt target genes [18,19]. Axin2 is a generic Wnt transcriptional target gene that is often used as an indicator of activation of this pathway [20].
2.2. The Noncanonical Pathways
Most studies have focused on the role of Wnt in the canonical signaling pathway; however, Wnt can also signal through a number of noncanonical pathways. These pathways include the Wnt-JNK, Wnt-RAP1, Wnt-Ror2, Wnt-PKA, Wnt-GSK3MT, Wnt-aPKC, Wnt-RYK, Wnt-mTOR, and Wnt/Ca2+ signaling pathways. Most of these pathways overlap to transduce calcium-dependent cell signaling [21]. It is thought that the transmembrane receptor Fz is also involved in these pathways, whereas the coreceptor LRP5/6 is thought to function only in canonical pathway signaling. In fact, there is in vivo evidence to suggest that LRP6 may antagonize noncanonical Wnt pathways, possibly to eliminate competition for Wnt ligands [22]. Ror1/2 is the coreceptor often involved in noncanonical pathways. Ligand interaction with this coreceptor causes an increase in inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 causes a release of Ca2+ from the endoplasmic reticulum, leading to activation of protein kinase C (PKC). PKC activates nuclear factor kappa-B (NFκB) and cAMP response element binding protein (CREB). Calcineurin (Cn) and calcium/calmodulin-dependent protein kinase type II (CamKII) are also activated, leading to activation of the nuclear factor of activated T cells (NFAT) and NFκB. NFκB, CREB, and NFAT translocate to the nucleus causing transcription of regulatory genes [21]. Although these many mechanisms may not be fully understood, it is clear that Wnt is involved in vital cellular functions, such as assisting in stabilization of proteins during mitosis, through these alternative pathways [23].
2.3. Wnt Inhibitors
There are several protein families and genes known to antagonize or modulate Wnt signaling. DKK1, a member of the Dickkopf (DKK) family of proteins, is thought to act as a LRP5/6 ligand antagonist [24] (Figure 1). There are different ideas on the mechanism of this action, with supporting evidence that DKK1 may induce LRP6 internalization and degradation or it may disrupt the Wnt-induced Fz-LRP6 complex [25]. Interestingly, while DKK1 was thought to be tumor suppressive in nature, it is now found to be associated with poor prognosis, supporting tumor growth and metastasis [25]. Contrarily, tumor immune evasion by Dickkopf-related protein 2 (DKK2), with LRP5, is thought to act independently of the Wnt/β-catenin pathway, via inhibition of STAT5 signaling [26]. Sclerostin/SOST proteins are secreted FZD-related proteins. These and Wnt Inhibitory Protein also act as inhibitors by direct interaction with Wnt [8]. Rnf43 and Znrf3 are two Wnt target genes that act as negative-feedback regulators, as they cause degradation of Wnt receptors [27,28,29]. Overall, the roles of these inhibitors and regulators need further investigation, as their ability to modulate abnormal Wnt may be therapeutically useful in a variety of disease settings.
3. Wnt/β-Catenin Signaling and Immunomodulation
3.1. Stem Cells
The Wnt signaling pathway is known to be involved in regulating the self-renewal capacity of non-malignant stem cells. For example, an association between Wnt and adult stem cells was established with the gene disruption of mouse TCF7L2. This gene encodes for T cell factor-4 (Tcf-4), which forms the β-catenin/Tcf-4 transcription complex. A study by Korinek et al. demonstrated that cessation of Wnt signaling resulted in a loss of intestinal stem cells, leading to a breakdown of the intestinal epithelium [30]. Wnt signaling has also been found to help maintain the pluripotency of embryonic stem cells [31]. In contrast, DKK1 overexpression, which results in Wnt pathway inhibition, has been shown to eliminate hair follicles and other skin appendages, suggesting a possible blockade of stem cell initiation [32]. Intriguingly, many studies are now analyzing tumor stem cells as a source of immune evasion, as they have been shown to selectively acquire expression of CD80, a surface ligand that dampens immune recognition by binding Cytotoxic T lymphocyte Antigen-4 (CTLA-4) present on activated T cells [33]. Overexpression of WNT5A, a common Wnt ligand, via epigenetic activation in glioblastoma is thought to lead to stem cell differentiation and invasive growth [34]. Additionally, epithelial-to-mesenchymal transition (EMT) and metastasis are supported by WNT5A [35]. In addition, mammary tumor stem cells were found to rely on Wnt proteins as rate-limiting self-renewal signals [36]. There is evidence to support Wnt pathway inhibition with the downregulation of PD-L1 expression, associated with a decreased stemness score signature, in triple negative breast cancer [37]. Potential relationships between cancer stem cells and CD8+ tumor infiltration, as related to tumor PD-L1 expression and the effects on cancer progression, are also being investigated [38]. Due to these documented effects of Wnt signaling on promoting stem cell viability and function, it is possible that aberrant Wnt signaling pathways may promote the stem-cell-like qualities of tumor stem cells, thereby facilitating intratumoral immune evasion.
With its role in cellular regulation, there is ample evidence that Wnt signaling affects hematopoietic stem cells. Wnt stimulation of hematopoietic stem cells may increase their self-renewal capacity [39]. For example, limiting β-catenin activation via noncanonical Wnt signaling stimulation was shown to inhibit the differentiation of hematopoietic stem cells [40,41]. However, other animal studies where β-catenin was mutated did not show a significant change in hematopoiesis [42,43]. Induction of the Wnt/β-catenin pathway through inhibition of GSK3β or the use of WNT3A, a stimulating Wnt ligand, was found to arrest CD8+ T cell differentiation into effector cells, promoting self-renewing multipotent CD8+ memory stem cells and maintaining the “stemness” of mature memory CD8+ T cells [44]. This correlation at the cellular developmental level may provide one of the key links between Wnt signaling and alterations in immune functionality in the tumor.
3.2. Wnt Signaling and Cancer
The associations between Wnt signaling and cancer progression are the topic of intense investigation. This correlation was first established when a WNT factor gene was identified as an oncogene in the mouse mammary cell line RAC311c [45]. Our understanding of these associations has now progressed to include the well-established relationship between APC gene mutations and colorectal cancers. APC gene mutations can be found in most sporadic colorectal cancers, and familial adenomatous polyposis, or the hereditary colon cancer syndrome [46,47]. Alterations in core Wnt regulators were found in a sequencing project of 1134 colorectal cancer samples, noting the incidence of oncogenic Wnt activation in 96% of human colorectal cancers [48]. Axin2 gene mutations have been found in colorectal cancers as well [49]. Hepatocellular carcinomas were found to have Axin1 mutations [50]. Deletions of GSK3B may lead hematopoietic stem cells to progress to acute myeloid leukemia [51]. Mutations in these proteins cause a stabilization of cytoplasmic β-catenin due to an inappropriately functioning degradation complex, thus mimicking an upregulation of Wnt signaling. Furthermore, direct mutations in β-catenin have been found in colon cancer and melanoma [52,53]. It is known that β-catenin is also used in cellular adhesion junctions. Through immunohistochemistry evaluation of epithelial ovarian cancer samples, a build-up of β-catenin in the cellular membrane was associated with a decrease in progression-free survival, and resistance to platinum-based chemotherapy [54]. Inactivating gene mutations in the pathway, such as Rnf43 in pancreatic cancer and Znrf3 in adrenocortical cancer or additional cancers, have implicated new links between Wnt signaling and cancer transformation [55,56]. While these direct changes of the Wnt pathway have been established in cancer progression, evolving investigations suggest more depth to Wnt’s role, specifically in regulating anti-tumor immunity.
3.3. Immunity in Cancer
Despite tremendous advancements in traditional chemotherapies, many oncology patients continue to experience rapid progression of disease characterized by chemotherapy resistance, which limits therapeutic options. This has led to the pressing need to investigate alternative methods of treatment, such as immune-directed therapies. Many key aspects of the relationship between the immune system and solid tumors have been elucidated over the last 20 years. There is an appreciable complexity of this system that results in either tumor suppression or tumor progression. It is now established that in order for immune cells to recognize cancer cells and control cancer progression and metastasis, they must first infiltrate the tumor, then remain activated in the TME [57]. The presence and activation of these tumor infiltrating lymphocytes (TILs) typically correlates with tumors that are more sensitive to chemotherapeutic treatment [58,59]. However, if the tumor has been invaded by tumor-promoting T cells, such as Tregs, there may be decreased treatment sensitivity [60]. In particular, higher frequencies of CD8+ T cells, and high CD8+/CD4+ ratios have been correlated with improved overall survival in ovarian cancer [61]. The main goal of immunotherapy is to convert the tumor milieu from an immunologically suppressed state to an inflamed state, for tumor recognition, cell destruction, and improved treatment sensitivity.
The Cancer Genome Atlas (TCGA) has provided much insight into the role of the immune system in various types of cancers through combined analysis of genomic and patient outcome data. With this resource, TME may be examined from a transcriptional viewpoint, permitting more nuanced cancer categorizations to be made. For example, Thorsson et al. recently performed an immunogenomic analysis of 10,000 tumors from 33 cancer types to identify underlying immune subtypes that are common to all cancers examined [62]. The six subtypes, namely wound healing, interferon-γ dominant, inflammatory, lymphocyte depleted, immunologically quiet and transforming growth factor beta (TGF-β) dominant, help identify the immunological differences present in TME signatures. From this and similar studies, future therapies may be more directed toward the appropriate targets.
3.4. Wnt Signaling and Leukocyte Differentiation
The differentiation of multiple leukocyte populations is regulated by Wnt signaling pathways. Alterations in canonical Wnt signaling may have a genomic influence on T cell development. TCF1 and LEF1 genes have been linked to epigenetic changes that may promote CD8+ T cell differentiation by repressing CD4 genetic networks [63,64]. Furthermore, deletions of Ctnnb1, the gene encoding for β-catenin, or deletions in Tcf7, the gene encoding TCF1, were shown to block thymocyte development [65,66]. When Tcf7 is deleted in CD8+ T cells, functional T cell memory is impaired [67]. However, when p45, a TCF1 variant, is combined with stabilized β-catenin, there is an enhancement of central memory T cell production [68]. Thus, there is support that genomic Wnt pathway alterations correlate with changes in T cell development and differentiation.
Other immune cell lineages are also influenced by canonical Wnt signaling. Innate lymphoid cells, including natural killer (NK) cells, require TCF1 for development, as shown by the earliest linage-specific precursor expressing high levels of TCF1, and defective NK cell survival in Tcf7-/- mice [69,70]. In Lef1-/- and Fzd9-/- mice, B cell precursors were diminished in the bone marrow [71]. However, B cell development was not impaired despite a Ctnnb1 deletion in B cell precursors [72]. Furthermore, dendritic cell (DC) differentiation is promoted when there is an upregulation of Fz receptors in DC precursors [73]. The observed results of Wnt signaling alterations throughout multiple cell types remains vast, reflecting the importance of this pathway in immune cellular regulation.
3.5. Wnt Signaling and Immunomodulation
Multiple leukocyte populations within the TME are influenced by both Wnt stimulators and inhibitors. Key Wnt-related immunological alterations are illustrated in Figure 2. For example, the Wnt/β-catenin inhibitor DKK1 has been found to be overexpressed in several different TMEs. High levels of DKK1 were found in serum samples of patients with pancreas, stomach, liver, bile duct, breast, and cervical carcinoma [74]. One might speculate that this could be a negative feedback result of deregulated Wnt signaling from the tumor. However, DKK1 binding to its receptor cytoskeleton associated protein 4 (CKAP4) promoted tumor progression [75]. Additional studies have shown tumor stroma-derived DKK1 targeted β-catenin downregulation in myeloid-derived suppressor cells (MDSCs), leading to an accumulation of these cells, a suppressed T cell response, and tumor proliferation [76]. When a DKK1 vaccination was given in a murine model of myeloma, it was shown to elicit CD4+ and CD8+ T cell protective immunity [77]. This insinuates a potential for a DKK1 vaccination as an immunotherapeutic adjunct. While the relationship between malignancy and DKK1 remains unclear, there appears to be a strong immunologic influence.
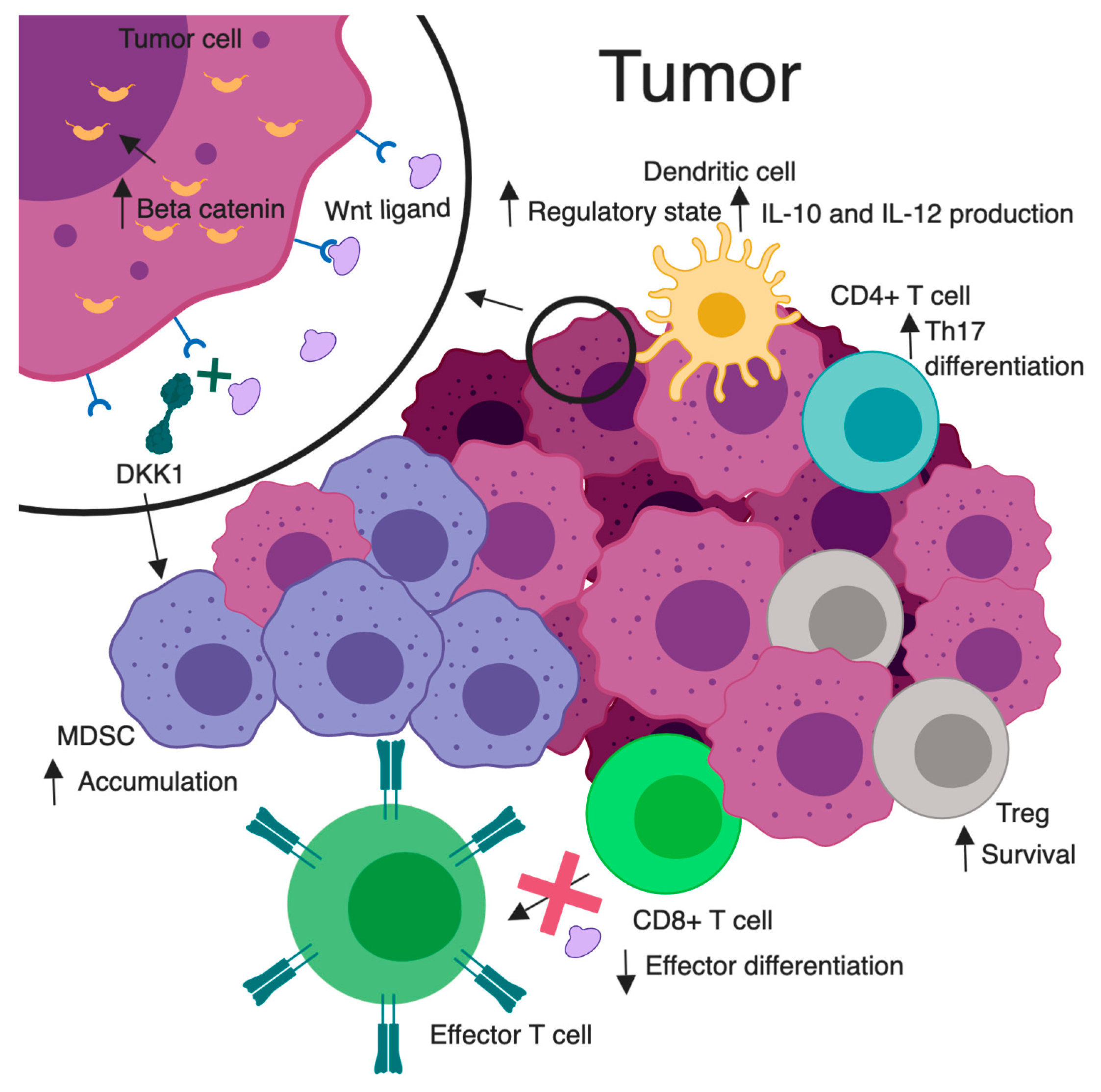
Figure 2. Wnt/β-catenin signaling and tumor immunomodulation. A magnified tumor cell (top left), illustrates Wnt signaling leading to elevated cytosolic and nuclear β-catenin. Increased Wnt signaling is associated with heightened survival of Tregs, skewed differentiation of CD4+ T cells to a pro-tumorigenic Th17 subtype, conversion of dendritic cells to a regulatory state with enhanced IL-10 and IL-12 secretion, and decreased effector differentiation and function in CD8+ T cells. DKK1 inhibits Wnt ligand/receptor interactions. Elevated DKK1 leads to an accumulation of MDSC in the TME and subsequent inhibition of effector CD8+ T cell function. Created with BioRender.com. Not drawn to scale.
Noncanonical pathway stimulation may support a tumor proliferative environment. WNT5A stimulation has been shown to increase IL-12 production from DCs, causing an increase in TH1 responses [78]. Alternatively, a deficiency of WNT5A in another model showed low levels of interferon gamma producing TH1 cells [79]. These Wnt effects on peripheral T cells may alter their functions in tumor recognition. For example, RAR-related orphan receptor C (RARC) was upregulated when CD4+ T cells had sustained β-catenin activation, resulting in TH17 polarization and production of proinflammatory cytokines that favor tumorigenesis [80]. It has also been shown that Treg survival is increased with increased β-catenin expression [81]. These recognized changes in tumor-infiltrating leukocytes provide insight into the influence of Wnt pathways on tumor immunity and provide a platform for new intervention concepts.
Immune tolerance is also impacted by Wnt signaling. Irregularities in antitumor cytotoxic T lymphocyte (CTL) priming are associated with Wnt signaling in DCs. The high levels of Wnt ligands found in the TME condition DCs to a regulatory state [82] (Figure 2). This suppressed antitumor immunity was explored via DC-specific LRP5/6 deletions in a murine tumor model. Results showed delayed tumor growth with enhanced effector T cell differentiation, and decreased Treg differentiation [83]. This idea was mimicked pharmacologically via use of the PORCN inhibitor IWP-L6 [83]. In one study, denilukin diftitox (ONTAK; a diphtheria toxin fragment/ IL-2 fusion protein) was given prior to DC vaccinations in patients with melanoma. This resulted in increased β-catenin in the skin and immune tolerance with an increased survival of resting Tregs [84]. In another study, forced expression of non-degradable β-catenin in melanoma cells or DCs led to secretion of the anti-inflammatory cytokine IL-10, which impaired the ability of DCs to cross-prime CD8+ CTLs for tumor recognition [85,86]. Through these influences on DCs, related to tumor-induced increases in β-catenin signaling, tumors may acquire tolerogenic characteristics, allowing immune evasion. However, in DC-β-catenin-/- mice, with a CD11c-specific deletion of β-catenin, vaccination with tumor antigen failed to provide tumor protection. Interestingly, these mice were found to be deficient in CD8+ T cell immunity [86]. In a mouse model of human melanoma, the β-catenin-dependent transcription blocker, PKF115-584, stimulates DCs to cross-prime tumor-specific CTLs, altering Wnt-induced immunosuppression and improving therapeutic response [85]. These findings suggest complex roles for β-catenin in DCs, where an aberrant amount, through either depletion or overabundance, may lead to immune tolerance.
Evidence suggests a strong correlation between Wnt pathway changes and immune exclusion. This was recently evaluated with TCGA data. Multiple cancers were analyzed for gene expression of TILs and categorized into a high, intermediate, or low T cell-inflamed tumor environment, based on their expression for genes associated with T cell infiltration. These tumors were then profiled for Wnt/β-catenin-related gene expression profiles. Up to 90% of tumor types showed an inverse correlation between Wnt/β-catenin pathway activation and a T cell-inflamed gene expression signature [87], suggesting that Wnt signaling in the TME suppresses T cell infiltration and/or function. Additional support of immune exclusion was found through a mouse melanoma model that was engineered to express β-catenin. These cells were unable to express C-C motif chemokine ligand 4 (CCL4), leading to decreased CTL infiltration into the TME due to defective recruitment of DCs [88]. Furthermore, in patients with primary and metastatic melanomas treated with BRAF inhibitors, tumor immune infiltration and survival were inversely correlated with β-catenin signaling [89,90]. Collectively, these findings imply a role for altered β-catenin levels in the exclusion of TILs in the TME.
Immunoevasion mechanisms may also be represented through alterations in immune checkpoint molecules due to Wnt signaling component changes. GSK3 inactivation in mouse melanoma resulted in a repression of the PD-1 gene, allowing an improved CD8+ response [91]. However, in mouse mammary carcinoma, GSK3β was shown to interact with PD-L1, inducing its degradation, which led to increased CTL infiltration [92]. As immune therapies become increasingly important in cancer therapeutic options, these interactions need to be further investigated.
3.6. Immunotherapy
Some immunotherapies are directed at the proteins that regulate T cell function and cytolytic activity. In particular, immune checkpoint inhibitors (ICIs) are monoclonal antibodies against receptors such as CLTA-4 and Programmed Death-1 (PD-1) that act to down-modulate T cell effector function. ICI are now approved for the treatment of malignant melanoma, non-small-cell lung cancer, classical Hodgkin lymphoma, head and neck squamous cell carcinoma, urothelial carcinoma, and renal cell carcinoma [93]. In addition, anti-programmed death-1 ligand (PD-L1) has also shown many promising clinical results [94]. Atezolizumab is an FDA-approved PD-L1 inhibitor that has shown improved progression-free survival and overall survival when used in combination with nabpaclitaxel for metastatic triple negative breast cancer [95]. These results were in patients with known positive PD-L1 tumors. Other anti-PD-L1 therapies include durvalumab and avelumab. Additionally, the monoclonal antibody pembrolizumab (anti-PD-1) was combined with platinum-based chemotherapy in metastatic non-small-cell lung cancer treatment in those who lacked targetable gene mutations. The combination of therapies increased progression-free and overall survival [96].
In a similar fashion, ipilimumab and tremelimumab are monoclonal antibodies to CTLA4, which normally functions to restrain effector T cell activation. Ipilimumab is known for its significant progression-free and overall survival advancement in melanoma [97]. Further improvements in metastatic melanoma were seen following a combination of ipilimumab and anti-PD-L1 therapy [98].
With the rising use of mono- and combinatorial immunotherapy in the clinical setting, there is a need to further understand the TME in patients who do not respond. It has been reported that decreased TILs result in resistance to ICIs [99]. Wnt pathway regulation may play a role in this lack of response, as tumor-intrinsic, active β-catenin signaling in human melanoma has been associated with resistance to anti-CTLA4 and anti-PD-L1 antibodies due to T cell exclusion [88]. It has also been speculated that tumor cells may lack the antigens needed for recognition by TILs, perhaps leading to immunotherapy resistance in these cancers. This was investigated with an analysis of 266 melanoma tumor samples from TCGA. Tissues were divided into categories based on a high or low expression of genes that were associated with T cell infiltration. This was correlated with nonsynonymous somatic mutations as a representative of mutational neoantigens that the tumor may possess. It was concluded that the change in tumor gene expression of infiltrating T cells did not correlate with increased mutational neoantigens [100]. However, gene signatures of these melanoma tissues did support prior evidence of a correlation between Wnt/β-catenin pathway activation and a reduction in T cell infiltration in the tumor [100]. This evidence, in addition to prior stated support, shows a strong correlation between increased Wnt signaling and decreased T cell infiltration in tumors.
4. Targeting Wnt Signaling as a Novel Therapeutic Option
There are numerous relationships between Wnt signaling, immune function, and cancer progression; most notable is that of increased Wnt signaling correlating with decreased tumor T cell infiltration, as discussed above. Wnt pathways have a vast number of roles that offer multiple options for pathway modulation in the malignant setting. The United States Patent and Trade Office Patent and Patent Application databases report 103 unique Wnt signaling modulators being investigated, with 34 in clinic trials [101]. Actions of these therapies range from the signal pathway component targeting activators to Wnt inhibitors. Many intriguing investigations are evaluating tumor immunity with drug intervention. Understanding the immunomodulation of these therapies will be essential during attempts to transition the TME to a less resistant milieu.
Numerous studies are currently combining Wnt therapies with ICIs, additional Wnt inhibitors, or chemotherapies in an attempt to achieve optimal effects on tumor control. However, it is currently unknown how the combination of therapies will affect tumor progression. In colon cancer, changes in Wnt signaling pathways have shown counter-intuitive results in some studies, such as WNT-TCF blockade actually boosting metastasis [102]. In contrast, other results have suggested a synergy between Wnt pathway inhibition and ICIs, for example when used as a combinatorial therapy in a mouse melanoma model [103]. Additional combination treatments with multiple Wnt inhibitors have been found to revert resistance and repress tumor growth in colorectal cancer [104]. Furthermore, the combination of Wnt antagonists with taxane therapies elicited a synergistic effect by sensitizing cancer stem cells to taxane-induced death [105]. Table 1 identifies clinical trials of Wnt modulators, with the listed agent, mechanism of action, intervention strategy, and targeted disease.
Table 1. Cancer Clinical Trials of Wnt Modulators. Agents used are listed with the mechanism of action, the corresponding investigated disease, the phase of the trial, the investigational intervention, and the clinical trial identifier [106].
| Agent | Mechanism of Action | Disease | Phase | Therapy | Identifier |
|---|---|---|---|---|---|
| DKN-01 | DKK1 antibody | Esophageal Neoplasms | 1 | Dose escalation in combination with paclitaxel or pembrolizumab | NCT02013154 |
| Adenocarcinoma of the Gastroesophageal Junction | |||||
| Gastroesophageal Cancer | |||||
| Squamous Cell Carcinoma | |||||
| Gastric Adenocarcinoma | |||||
| DKN-01 | DKK1 antibody | Endometrial Cancer | 2 | Monotherapy or in combination with paclitaxel | NCT03395080 |
| Uterine Cancer | |||||
| Ovarian Cancer | |||||
| DKN-01 | DKK1 antibody | Hepatocellular Carcinoma | 1, 2 | Phase 1/2 as a monotherapy or combination with sorafenib | NCT03645980 |
| DKN-01 | DKK1 antibody | Multiple Myeloma | 1 | Pilot study of combination with lenalidomide/dexamethasone | NCT01711671 |
| DKN-01 | DKK1 antibody | Multiple Myeloma | 1 | Dose escalation | NCT01457417 |
| Solid Tumors | |||||
| Non-Small Cell Lung Cancer | |||||
| DKN-01 | DKK1 antibody | Carcinoma of Intrahepatic and Extra-hepatic Biliary System | 1 | Dose escalation combined with gemcitabine and cisplatin | NCT02375880 |
| Carcinoma of Gallbladder | |||||
| Bile Duct Cancer | |||||
| Cholangiocarcinoma | |||||
| CGX 1321 | PORCN inhibitor | Colorectal Adenocarcinoma | 1 | Single agent dose escalation | NCT03507998 |
| Gastric Adenocarcinoma | |||||
| Pancreatic Adenocarcinoma | |||||
| Bile Duct Carcinoma | |||||
| Hepatocellular Carcinoma | |||||
| Esophageal Carcinoma | |||||
| Gastrointestinal Cancer | |||||
| CGX 1321 | PORCN inhibitor | Solid Tumors | 1 | Single agent dose escalation with or without pembrolizumab | NCT02675946 |
| GI Cancer | |||||
| ETC1922159 | PORCN inhibitor | Solid Tumors | 1 | Single agent dose escalation | NCT02521844 |
| LGK974 | PORCN inhibitor | Pancreatic Cancer | 1 | Single agent and in combination with PDR001 | NCT01351103 |
| BRAF Mutant Colorectal Cancer | |||||
| Melanoma | |||||
| Triple Negative Breast Cancer | |||||
| Head and Neck Squamous Cell Cancer | |||||
| Cervical Squamous Cell Cancer | |||||
| Esophageal Squamous Cell Cancer | |||||
| Lung Squamous Cell Cancer | |||||
| RXC004 | PORCN inhibitor | Cancer | 1 | Dose tolerability | NCT03447470 |
| Solid Tumor | |||||
| Artesunate | Unknown | Hepatocellular Carcinoma | 1 | Single agent dose escalation | NCT02304289 |
| Artesunate | Unknown | Colorectal Cancer | 2 | Neoadjuvant single agent | NCT03093129 |
| Artesunate | Unknown | Solid Tumors | 1 | Single agent dose escalation | NCT02353026 |
| Artesunate | Unknown | Colorectal Cancer | 2 | Neoadjuvant single agent | NCT02633098 |
| Bowel Cancer | |||||
| Artesunate | Unknown | Metastatic Breast Cancer | 1 | Add-on therapy | NCT00764036 |
| Locally Advanced Breast Cancer | |||||
| Niclosamide | AXIN1 activator | Colon Cancer | 1 | Dose escalation | NCT02687009 |
| Niclosamide | AXIN1 activator | Metastatic Prostate Carcinoma | 1 | Dose escalation with enzalutamide | NCT03123978 |
| Recurrent Prostate Carcinoma | |||||
| Stage IV Prostate Cancer | |||||
| Niclosamide | AXIN1 activator | Castration-Resistant Prostate Carcinoma | 1 | Dose escalation with enzalutamide | NCT02532114 |
| Metastatic Prostate Carcinoma | |||||
| Recurrent Prostate Carcinoma | |||||
| Stage IV Prostate Adenocarcinoma | |||||
| Niclosamide | AXIN1 activator | Colorectal Cancer | 2 | Single agent | NCT02519582 |
| Niclosamide | AXIN1 activator | Metastatic Prostate Cancer | 2 | Combination with abirateronae acetate and prednisone | NCT02807805 |
| Recurrent Prostate Cancer | |||||
| Stage IV Prostate Cancer | |||||
| OMP54F28 | Wnt receptor decoy | Hepatocellular Cancer | 1 | Dose escalation with sorafenib | NCT02069145 |
| Liver Cancer | |||||
| OMP54F28 | Wnt receptor decoy | Ovarian Cancer | 1 | Combined with paclitaxel and carboplatin | NCT02092363 |
| OMP54F28 | Wnt receptor decoy | Pancreatic Cancer | 1 | Combined with Nab-paclitaxel and gemcitabine | NCT02050178 |
| Stage IV Pancreatic Cancer | |||||
| OMP54F28 | Wnt receptor decoy | Solid Tumors | 1 | Dose escalation | NCT01608867 |
| Foxy-5 | WNT5A mimic | Metastatic Breast Cancer | 1 | Dose escalation | NCT02020291 |
| Colorectal Cancer | |||||
| Prostate Cancer | |||||
| Foxy-5 | WNT5A mimic | Metastatic Breast Cancer | 1 | Dose escalation | NCT02655952 |
| Metastatic Colon Cancer | |||||
| Metastatic Prostate Cancer | |||||
| PRI724 | CBP/catenin inhibitor | Advanced Pancreatic Cancer | 1 | Dose escalation with gemcitabine | NCT01764477 |
| Metastatic Pancreatic Cancer | |||||
| Pancreatic Adenocarcinoma | |||||
| PRI724 | CBP/catenin inhibitor | Acute Myeloid Leukemia | 1, 2 | Dose escalation, combined with dasatinib for CML or cytarabine for AML | NCT01606579 |
| Chronic Myeloid Leukemia | |||||
| SM08502 | Unknown | Solid Tumors, Adult | 1 | Single agent dose escalation | NCT03355066 |
Therapeutic modulation of Wnt signaling is being investigated through many avenues, several of which are depicted in Figure 1. One agent under investigation is DKN-01. This is a monoclonal antibody to DKK1, the Wnt/β-catenin inhibitor. DKN-01 is being used in several clinical trials for investigation of safety and efficacy in patients with multiple primary tumor types. There have been two completed clinical trials with this drug in multiple myeloma (NCT01711671, NCT01457417). Results are available from one of these studies, but published conclusions are pending [106]. High serum levels of DKK1 were found in patients with pancreas, stomach, liver, bile duct, breast, and cervical cancers [74]. Prior studies have also shown that increased DKK1 stabilized MDSC populations, leading to suppression of the T cell intratumoral response [76]. The direct influence on Wnt signaling from these agents is convoluted given it is a result of inhibition of a Wnt inhibitor. If this inhibitor is blocking canonical Wnt signaling, it may lead to an upregulation of noncanonical signaling. Perhaps with inhibition of DKK1, the alternative pathway will be normalized. There may also be a decrease in intratumoral suppressive MDSCs, resulting in increased tumor cell recognition and clearance by CD8+ T cells. It will be interesting to view future clinical trial results related to effects on the TME that occur following targeted inhibition of a Wnt inhibitor, with and without combination therapy.
Alternative therapeutic agents act directly on Wnt ligand secretion. PORCN inhibitors are known to block the extracellular excretion of Wnt by blocking the enzyme responsible for palmitoylation of Wnt ligands (Figure 1). This family of inhibitors includes C59, CGX 1321, ETC1922159, LGK974, IWP-L6, and RXC004. With this overall extracellular decrease in Wnt ligands, the TME may have the ability to convert to a T cell-inflamed environment, based on evidence of increased Wnt signaling correlated with T cell-noninflamed tumors [87]. Many of these molecules remain under investigation in the preclinical setting; however, some studies have advanced to clinical trials. Pending results will provide insight into treatment efficacy for multiple malignancies.
Some Wnt altering agents have been previously approved in nonmalignant diseases. Artesunate is a compound extracted from the herb Artemisia annua, used as an FDA-approved antimalarial drug. Treatment of colorectal tumor xenografts with this agent correlated with decreased growth of tumors with inhibition of a hyperactive Wnt/β-catenin pathway [107]. The exact mechanism of the agent remains unknown. However, two phase 1 trials are now completed using this agent in subjects with hepatocellular carcinoma or solid tumors. Results are pending from these dose-escalation studies (NCT02304289, NCT02353026). An additional completed phase 1 study evaluated artesunate as an add-on therapy in subjects with metastatic or locally advanced breast cancer, with unreleased results (NCT00764036). Additionally, niclosamide is an anti-helminthic agent that has been identified to have many molecular targets, including inhibition of the Wnt pathway. Specifically, the Axin-GSK3β interaction is targeted in this pathway, resulting in a suppression of Wnt/Snail [108]. There are several phase 1 trials involving this therapeutic agent in various cancers. One trial involving several types of prostate cancer has been completed, with pending results (NCT02532114). One note of caution is that with so many known targets, it may be hard to determine if the effects of these therapies are directly related to Wnt changes in the tumor, as opposed to additional mechanistic alterations.
Additional Wnt-inhibiting agents are being tested in clinical trials. Ipafricept, also known as OMP54F28, is a recombinant fusion protein with an extracellular Fzd 8 receptor portion attached to an IgG1 Fc fragment, which acts as a decoy receptor for Wnt ligands [109]. Four phase 1 trials have been completed with this therapy (NCT02069145, NCT02092363, NCT02050178, NCT01608867). Study conclusions have not been released. WNT5A is a Wnt ligand mimicked by Foxy-5, a formylated 6 amino acid peptide fragment. The agent is thought to impair migration of epithelial cancer cells, giving it anti-metastatic potential [110]. Two clinical trials have been completed to determine appropriate doses for phase 2 trials (NCT02020291, NCT02655952). Due to increased β-catenin levels found in many colon cancers, a CREB-binding protein (CBP)/catenin inhibitor, PRI724, is being investigated [111]. Two phase 1 clinical trials have been completed with the use of this inhibitor in pancreatic cancers and acute and chronic myeloid leukemias (NCT01764477, NCT01606579). Results from these clinical trials are currently unavailable. SM08502 is an orally bioavailable small molecule inhibitor that is thought to inhibit the expression of Wnt signaling pathway genes, but further investigation is being elucidated on the exact mechanisms of action and its relation to Wnt. One phase 1 clinical trial is using this agent in solid tumors (NCT03355066). Completion of these studies, and future studies, may provide insight into the optimal dose and timing for administration of Wnt-based therapeutics and malignancies that are most sensitive to these agents.
References
- Wang, B.; Tian, T.; Kalland, K.-H.; Ke, X.; Qu, Y. Targeting Wnt/β-Catenin Signaling for Cancer Immunotherapy. Trends Pharmacol. Sci. 2018, 39, 648–658.
- Galluzzi, L.; Spranger, S.; Fuchs, E.; López-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65.
- Mora, J.; Mertens, C.; Meier, J.K.; Fuhrmann, D.C.; Brüne, B.; Jung, M. Strategies to Interfere with Tumor Metabolism through the Interplay of Innate and Adaptive Immunity. Cells 2019, 8, 8.
- Sherwood, V. Wnt signaling: An emerging mediator of cancer cell metabolism? Cell. Biol. 2015, 35, 2–10.
- Roel Nusse, H.C. Wnt/b-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999.
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109.
- Kusserow, A.; Sturm, C.; Hrouda, M.; Lentfer, J.; Schmidt, H.A.; Technau, U.; von Haeseler, A.; Hobmayer, B.; Martindale, M.Q.; Holstein, T.W. Unexpected complexity of the wnt gene familly in a sea anemone. Nature 2005, 433, 156–160.
- Niehrs, C.; Acebron, S.P. Mitotic and mitogenic Wnt signalling. EMBO J. 2012, 31, 2705–2713.
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081.
- Janda, C.Y.; Waghray, D.; Levin, A.M.; Thomas, C.; Garcia, K.C. Structural basis of Wnt recognition by Frizzled. Science 2012, 337, 59–64.
- Bartscherer, K.; Pelte, N.; Ingelfinger, D.; Boutros, M. Secretion of Wnt Ligands Requires Evi, a Conserved Transmembrane Protein. Cell 2006, 125, 523–533.
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nature 2012, 14, 1036–1045.
- Alok, A.; Lei, Z.; Jagannathan, N.S.; Kaur, S.; Harmston, N.; Rozen, S.G.; Tucker-Kellogg, L.; Virshup, D.M. Wnt proteins synergize to activate b-catenin signaling. Cell Sci. 2017, 130, 1532–1544.
- Janda, C.Y.; Dang, L.T.; You, C.; Chang, J.; de Lau, W.; Zhong, Z.A.; Yan, K.S.; Marecic, O.; Siepe, D.; Li, X.; et al. Surrogate wnt agonists that phenocopy canonical wnt and b-catenin signaling. Nature 2017, 545, 234–237.
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804.
- Kitagawa, M.; Hatakeyama, S.; Shirane, M.; Matsumoto, M.; Ishida, N.; Hattori, K.; Nakamichi, I.; Kikuchi, A.; Nakayama, K.; Nakayama, K.-I.; et al. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999, 18, 2401–2410.
- Stamos, J.L.; Enos, M.D.; Shah, N.; Weis, W.I. Structural basis of gsk-3 inhibition by n-terminal phosphorylation and by the wnt receptor lrp6. eLife 2014, 3, e01998.
- Behrens, J.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor lef-1. Nature 1996, 382, 638–642.
- Molenaar, M.; Van De Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399.
- Lustig, B.; Jerchow, B.; Sachs, M.; Weiler, S.; Pietsch, T.; Karsten, U.; Van De Wetering, M.; Clevers, H.; Schlag, P.M.; Birchmeier, W.; et al. Negative Feedback Loop of Wnt Signaling through Upregulation of Conductin/Axin2 in Colorectal and Liver Tumors. Cell. Biol. 2002, 22, 1184–1193.
- De, A. Wnt/ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756.
- Bryja, V.; Andersson, E.R.; Schambony, A.; Esner, M.; Bryjová, L.; Biris, K.K.; Hall, A.C.; Kraft, B.; Cajanek, L.; Yamaguchi, T.P.; et al. The Extracellular Domain of Lrp5/6 Inhibits Noncanonical Wnt Signaling In Vivo. Biol. Cell 2009, 20, 924–936.
- Stolz, A.; Neufeld, K.; Ertych, N.; Bastians, H. Wnt-mediated protein stabilization ensures proper mitotic microtubule assembly and chromosome segregation. EMBO Rep. 2015, 16, 490–499.
- Cruciat, C.M. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081.
- Kagey, M.H.; He, X. Rationale for targeting the Wnt signalling modulator Dickkopf‐1 for oncology. J. Pharmacol. 2017, 174, 4637–4650.
- Xiao, Q.; Wang, W.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; Bosenberg, M.; et al. Dkk2 imparts tumor immunity evasion through β-catenin-independent suppression of cytotoxic immune-cell activation. Med. 2018, 24, 262–270.
- Hao, H.-X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200.
- Koo, B.-K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; Van De Wetering, M.; Van Es, J.H.; Mohammed, S.; Heck, A.J.R.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669.
- Moffat, L.L.; Robinson, R.E.; Bakoulis, A.; Clark, S.G. The conserved transmembrane RING finger protein PLR-1 downregulates Wnt signaling by reducing Frizzled, Ror and Ryk cell-surface levels in elegans. Development 2014, 141, 617–628.
- Korinek, V.; Barker, N.; Moerer, P.; Van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Genet. 1998, 19, 379–383.
- Berge, D.T.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nature 2011, 13, 1070–1075.
- Andl, T.; Reddy, S.T.; Gaddapara, T.; E Millar, S. WNT signals are required for the initiation of hair follicle development. F1000 Post-Publ. Peer Rev. Biomed. Lit. 2002, 2, 643–653.
- Miao, Y.; Yang, H.; Levorse, J.; Yuan, S.; Polak, L.; Sribour, M.; Singh, B.; Rosenblum, M.D.; Fuchs, E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 2019, 177, 1172–1186.e14.
- Hu, B.; Wang, Q.; Wang, Y.A.; Hua, S.; Sauvé, C.-E.G.; Ong, D.; Lan, Z.D.; Chang, Q.; Ho, Y.W.; Monasterio, M.M.; et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 2016, 167, 1281–1295.e18.
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856.
- Zeng, Y.A.; Nusse, R. Wnt Proteins Are Self-Renewal Factors for Mammary Stem Cells and Promote Their Long-Term Expansion in Culture. Cell Stem Cell 2010, 6, 568–577.
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060.
- Hou, Y.C.; Hsieh, M.H.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Low cd8⁺ t cell infiltration and high pd-l1 expression are associated with level of cd44⁺/cd133⁺ cancer stem cells and predict an unfavorable prognosis in pancreatic cancer. Cancers (Basel) 2019, 11, E541.
- Willert, K.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R., 3rd; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452.
- Sugimura, R.; He, X.C.; Venkatraman, A.; Arai, F.; Box, A.; Semerad, C.; Haug, J.S.; Peng, L.; Zhong, X.-B.; Suda, T.; et al. Noncanonical Wnt Signaling Maintains Hematopoietic Stem Cells in the Niche. Cell 2012, 150, 351–365.
- Nemeth, M.J.; Topol, L.; Anderson, S.M.; Yang, Y.; Bodine, D.M. Wnt5a inhibits canonical wnt signaling in hematopoietic stem cells and enhances repopulation. Natl. Acad. Sci. USA 2007, 104, 15436–15441.
- Cobas, M.; Wilson, A.; Ernst, B.; Mancini, S.J.; MacDonald, H.R.; Kemler, R.; Radtke, F. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. Exp. Med. 2004, 199, 221–229.
- Jeannet, G.; Scheller, M.; Scarpellino, L.; Duboux, S.; Gardiol, N.; Back, J.; Kuttler, F.; Malanchi, I.; Birchmeier, W.; Leutz, A.; et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood 2008, 111, 142–149.
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector t cell differentiation and generates cd8+ memory stem cells. Med. 2009, 15, 808–813.
- Rijsewijk, F.; Van Deemter, L.; Wagenaar, E.; Sonnenberg, A.; Nusse, R. Transfection of the int-1 mammary oncogene in cuboidal RAC mammary cell line results in morphological transformation and tumorigenicity. EMBO J. 1987, 6, 127–131.
- Kinzler, K.W. Lessions from hereditary colorectal cancer. Cell 1996, 87, 159–170.
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669.
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sánchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 33, 125–136.e3.
- Liu, W.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; Christensen, E.; et al. Mutations in axin2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/tcf signalling. Genet. 2000, 26, 146–147.
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Genet. 2000, 24, 245–250.
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74.
- Morin, P.J. Activation of beta-Catenin-Tcf Signaling in Colon Cancer by Mutations in beta-Catenin or APC. Science 1997, 275, 1787–1790.
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-Catenin by Genetic Defects in Melanoma Cell Lines. Science 1997, 275, 1790–1792.
- Bodnar, L.; Cierniak, S.; Smoter, M.; Cichowicz, M.; Kozlowski, W.; Szczylik, C.; Wieczorek, M.; Laparska-Przybysz, M. Wnt/b-catenin pathway as a potential prognostic and predictive marker in patients with advanced ovarian cancer. Ovarian Res. 2014, 7, 16.
- Wu, J.; Molin, M.D.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; Schulick, R.D.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrrent mutations in components of ubiquitin-dependent pathways. Natl. Acad. Sci. USA 2011, 108, 21188–21193.
- Assie, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Genet. 2014, 46, 607–612.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570.
- Maartje, C.A.; Wouters, F.L.K.; Workel, H.H.; Klip, H.G.; Plat, A.; Kooi, N.M.; Wisman, G.B.A.; Mourits, M.J.E.; Arts, H.J.G.; Oonk, M.H.M.; et al. Treatment regimen, surgical outcome, and t-cell differentiation influence prognostic benefit of tumor-infiltrating lymphocytes in high-grade serous ovarian cancer. Cancer Res. 2016, 22, 714–724.
- Ruan, M.; Tian, T.; Rao, J.; Xu, X.; Yu, B.; Yang, W.; Shui, R. Predictive value of tumor-infiltrating lymphocytes to pathological complete response in neoadjuvant treated triple-negative breast cancers. Pathol. 2018, 13, 66.
- Xiang, P.; Yang, Y.; Sheng, J.; He, Q.; Song, Y.; Yu, W.; Hu, S.; Jin, J. Infiltrating cd4+ t cells attenuate chemotherapy sensitivity in prostate cancer via ccl5 signaling. Prostate 2019, 79, 1018–1031.
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial cd8+ tumor-infiltrating lymphocytes and a high cd8+/regulatory t cell ratio are associated with favorable prognosis in ovarian cancer. Natl. Acad. Sci. USA 2005, 102, 18538–18543.
- Thorsson, V.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; Ziv, E.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.
- Xing, S.; Li, F.; Zeng, Z.; Zhao, Y.; Yu, S.; Shan, Q.; Li, Y.; Phillips, F.C.; Maina, P.K.; Qi, H.H.; et al. Tcf1 and lef1 transcription factors establish cd8(+) t cell identity through intrinsic hdac activity. Immunol. 2016, 17, 695–703.
- Steinke, F.C.; Yu, S.; Zhou, X.; He, B.; Yang, W.; Zhou, B.; Kawamoto, H.; Zhu, J.; Tan, K.; Xue, H.H. Tcf-1 and lef-1 act upstream of th-pok to promote the cd4(+) t cell fate and interact with runx3 to silence cd4 in cd8(+) t cells. Immunol. 2014, 15, 646–656.
- Staal, F.J.T.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Rev. Immunol. 2008, 8, 581–593.
- Xu, Y.; Banerjee, D.; Huelsken, J.; Birchmeier, W.; Sen, J.M. Deletion of beta-catenin impairs t cell development. Immunol. 2003, 4, 1177–1182.
- Jeannet, G.; Boudousquié, C.; Gardiol, N.; Kang, J.; Huelsken, J.; Held, W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Natl. Acad. Sci. USA 2010, 107, 9777–9782.
- Zhao, D.M.; Yu, S.; Zhou, X.; Haring, J.S.; Held, W.; Badovinac, V.P.; Harty, J.T.; Xue, H.H. Constitutive activation of wnt signaling favors generation of memory cd8 t cells. Immunol. 2010, 184, 1191–1199.
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/Planar Cell Polarity Signaling: Cellular Orientation by Facing the Wind (Wnt). Rev. Cell Dev. Biol. 2015, 31, 623–646.
- Jeevan-Raj, B.; Gehrig, J.; Charmoy, M.; Chennupati, V.; Grandclément, C.; Angelino, P.; Delorenzi, M.; Held, W. The Transcription Factor Tcf1 Contributes to Normal NK Cell Development and Function by Limiting the Expression of Granzymes. Cell Rep. 2017, 20, 613–626.
- Ranheim, E.A.; Kwan, H.C.K.; Wang, Y.-K.; Reya, T.; Weissman, I.L.; Francke, U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood 2005, 105, 2487–2494.
- Yu, Q.; Quinn, W.J., 3rd; Salay, T.; Crowley, J.E.; Cancro, M.P.; Sen, J.M. Role of beta-catenin in b cell development and function. Immunol. 2008, 181, 3777–3783.
- Zhou, J.; Cheng, P.; Youn, J.-I.; Cotter, M.J.; Gabrilovich, D.I. Notch and Wingless Signaling Cooperate in Regulation of Dendritic Cell Differentiation. Immunology 2009, 30, 845–859.
- Sato, N.; Yamabuki, T.; Takano, A.; Koinuma, J.; Aragaki, M.; Masuda, K.; Ishikawa, N.; Kohno, N.; Ito, H.; Miyamoto, M.; et al. Wnt Inhibitor Dickkopf-1 as a Target for Passive Cancer Immunotherapy. Cancer Res. 2010, 70, 5326–5336.
- Kimura, H.; Fumoto, K.; Shojima, K.; Nojima, S.; Osugi, Y.; Tomihara, H.; Eguchi, H.; Shintani, Y.; Endo, H.; Inoue, M.; et al. CKAP4 is a Dickkopf1 receptor and is involved in tumor progression. Clin. Investig. 2016, 126, 2689–2705.
- D’Amico, L.; Mahajan, S.; Capietto, A.-H.; Yang, Z.; Zamani, A.; Ricci, B.; Bumpass, D.B.; Meyer, M.; Su, X.; Wang-Gillam, A.; et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. Exp. Med. 2016, 213, 827–840.
- Qian, J.; Zheng, Y.; Zheng, C.; Wang, L.; Qin, H.; Hong, S.; Li, H.; Lu, Y.; He, J.; Yang, J.; et al. Active vaccination with dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012, 119, 161–169.
- Valencia, J.; Hidalgo, L.; Hernandez-Lopez, C.; Canseco, N.M.; Vicente, A.; Varas, A.; Sacedon, R. Wnt5a signaling increases il-12 secretion by human dendritic cells and enhances ifn-gamma production by cd4+ t cells. Lett. 2014, 162, 188–199.
- Sato, A.; Kayama, H.; Shojima, K.; Matsumoto, S.; Koyama, H.; Minami, Y.; Nojima, S.; Morii, E.; Honda, H.; Takeda, K.; et al. The wnt5a-ror2 axis promotes the signaling circuit between interleukin-12 and interferon-gamma in colitis. Rep. 2015, 5, 10536.
- Keerthivasan, S.; Aghajani, K.; Dose, M.; Molinero, L.; Khan, M.W.; Venkateswaran, V.; Weber, C.; Emmanuel, A.O.; Sun, T.; Bentrem, D.J.; et al. Beta-catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in t cells. Transl. Med. 2014, 6, 225ra228.
- Ding, Y.; Shen, S.; Lino, A.C.; Lafaille, M.A.C.D.; Lafaille, J.J. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Med. 2008, 14, 162–169.
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Majumdar, T.; Angus-Hill, M.L.; Koni, P.A.; Manicassamy, B.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. Beta-catenin promotes regulatory t-cell responses in tumors by inducing vitamin a metabolism in dendritic cells. Cancer 2015, 75, 656–665.
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Shanmugam, A.; Swafford, D.; Ahmad, S.; Chinnadurai, R.; Manicassamy, B.; He, Y.; Mellor, A.L.; et al. Deletion of lrp5 and lrp6 in dendritic cells enhances antitumor immunity. Oncoimmunology 2016, 5, e1115941.
- Baur, A.S.; Lutz, M.B.; Schierer, S.; Beltrame, L.; Theiner, G.; Zinser, E.; Ostalecki, C.; Heidkamp, G.; Haendle, I.; Erdmann, M.; et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood 2013, 122, 2185–2194.
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune suppression and resistance mediated by constitutive activation of wnt/beta-catenin signaling in human melanoma cells. Immunol. 2012, 189, 2110–2117.
- Fu, C.; Liang, X.; Cui, W.; Ober-Blobaum, J.L.; Vazzana, J.; Shrikant, P.A.; Lee, K.P.; Clausen, B.E.; Mellman, I.; Jiang, A. Beta-catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of cd8+ t cells through regulation of il-10. Natl. Acad. Sci. USA 2015, 112, 2823–2828.
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. Wnt/beta-catenin pathway activation correlates with immune exclusion across human cancers. Cancer Res. 2019, doi:10.1158/1078-0432.CCR-18-1942.
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235.
- Nsengimana, J.; Laye, J.; Filia, A.; O’Shea, S.; Muralidhar, S.; Pozniak, J.; Droop, A.; Chan, M.; Walker, C.; Parkinson, L.; et al. Beta-catenin-mediated immune evasion pathway frequently operates in primary cutaneous melanomas. Clin. Investig. 2018, 128, 2048–2063.
- Massi, D.; Romano, E.; Rulli, E.; Merelli, B.; Nassini, R.; De Logu, F.; Bieche, I.; Baroni, G.; Cattaneo, L.; Xue, G.; et al. Baseline beta-catenin, programmed death-ligand 1 expression and tumour-infiltrating lymphocytes predict response and poor prognosis in braf inhibitor-treated melanoma patients. J. Cancer 2017, 78, 70–81.
- Taylor, A.; Rothstein, D.; Rudd, C.E. Small-molecule inhibition of pd-1 transcription is an effective alternative to antibody blockade in cancer therapy. Cancer Res. 2018, 78, 706–717.
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Commun. 2016, 7, 12632.
- Simsek, M.; Bilici, M. Immunological agents used in cancer treatment. Eurasian J. Med. 2019, 1, 90–94.
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Rev. Drug Discov. 2016, 15, 235–247.
- Cortés, J.; André, F.; Gonçalves, A.; Kümmel, S.; Martín, M.; Schmid, P.; Schuetz, F.; Swain, S.M.; Easton, V.; Pollex, E.; et al. IMpassion132 Phase III trial: Atezolizumab and chemotherapy in early relapsing metastatic triple-negative breast cancer. Oncol. 2019, doi:10.2217/fon-2019-0059.
- Wang, C.; Kulkarni, P.; Salgia, R. Combined Checkpoint Inhibition and Chemotherapy: New Era of 1st-Line Treatment for Non-Small-Cell Lung Cancer. Ther. Oncolytics 2019, 13, 1–6.
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Cancer Res. 2019, doi:10.1158/1078-0432.CCR-18-1550.
- Wolchok, J.D.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; Smylie, M.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. Engl. J. Med. 2017, 377, 1345–1356.
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Rev. Mol. Cell Biol. 2018, 19, 731–745.
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Natl. Acad. Sci. USA 2016, 113, E7759–E7768.
- Lu, B.; Green, B.A.; Farr, J.M.; Lopes, F.C.; Van Raay, T.J.; Lo, H.-W. Wnt Drug Discovery: Weaving Through the Screens, Patents and Clinical Trials. Cell. Basis Metastasis Road Ther. 2016, 8, 82.
- Seth, C.; Altaba, A.R. Metastases and Colon Cancer Tumor Growth Display Divergent Responses to Modulation of Canonical WNT Signaling. PLoS ONE 2016, 11, e0150697.
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-derived wnt5a promotes local dendritic-cell expression of ido and immunotolerance: Opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095.
- Arqués, O.; Puig, I.; Tenbaum, S.P.; Argilés, G.; Dienstmann, R.; Fernández, N.; Caratù, G.; Matito, J.; Silberschmidt, D.; Rodon, J.; et al. Tankyrase inhibition blocks wnt/β-catenin pathway and reverts resistance to pi3k and akt inhibitors in the treatment of colorectal cancer. Cancer Res. 2016, 22, 644–656.
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.-Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Adv. 2017, 3, e1700090.
- S. National Library of Medicine. Clnicaltrial.Gove. Available online: https://clinicaltrials.gov/ct2/home (accessed on 30 January 2019).
- Li, L.N.; Zhang, H.D.; Yuan, S.J.; Tian, Z.Y.; Wang, L.; Sun, Z.X. Artesunate attenuates the growth of human colorectal carcinoma and inhibits hyperactive wnt/beta-catenin pathway. J. Cancer 2007, 121, 1360–1365.
- Ahn, S.Y.; Kim, N.H.; Lee, K.; Cha, Y.H.; Yang, J.H.; Cha, S.Y.; Cho, E.S.; Lee, Y.; Cha, J.S.; Cho, H.S.; et al. Niclosamide is a potential therapeutic for familial adenomatosis polyposis by disrupting Axin-GSK3 interaction. Oncotarget 2017, 8, 31842–31855.
- Jimeno, A.; Chugh, R.; Dupont, J.; Uttamsingh, S.; Kapoun, A.M.; Smith, D.C.; Messersmith, W.; Stagg, R.; Xu, L.; Brachmann, R.K.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Cancer Res. 2017, 23, 7490–7497.
- Canesin, G.; Evans-Axelsson, S.; Hellsten, R.; Krzyzanowska, A.; Prasad, C.P.; Bjartell, A.; Andersson, T. Treatment with the WNT5A-mimicking peptide Foxy-5 effectively reduces the metastatic spread of WNT5A-low prostate cancer cells in an orthotopic mouse model. PLoS ONE 2017, 12, e0184418.
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/creb-binding protein transcription [corrected]. Natl. Acad. Sci. USA 2004, 101, 12682–12687.


