 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elia Torres-Gutiérrez | -- | 2131 | 2023-02-28 15:33:10 | | | |
| 2 | Elia Torres-Gutiérrez | -139 word(s) | 1992 | 2023-03-01 21:46:27 | | | | |
| 3 | Dean Liu | + 23 word(s) | 2015 | 2023-03-02 03:28:20 | | |
Video Upload Options
Chagas disease is a complex zoonosis. Its natural history involves the interaction of transmitting arthropods with wild, peridomestic, and domestic mammals, and it has a great diversity of transmission forms. In a vertebrate host, the disease has two clinical phases: an acute phase and a chronic one; the former evolves without demonstrated pathology and can last 10–20 years. After this phase, some patients progress to the chronic symptomatic phase, in which they develop mainly cardiac lesions. The lesions in this cardiomyopathy involve several cardiac tissues, mainly the myocardium, and in severe cases, the endocardium pericardium; this can cause pleural effusion, which may evolve into sudden death, which is more frequent in cases with dilated heart disease and severe heart failure.
1. Introduction
2. Pathogenic Mechanisms of Immune Response Evasion in Chagas Disease
3. Histopathological Mechanisms Related to Tissue Parasitism
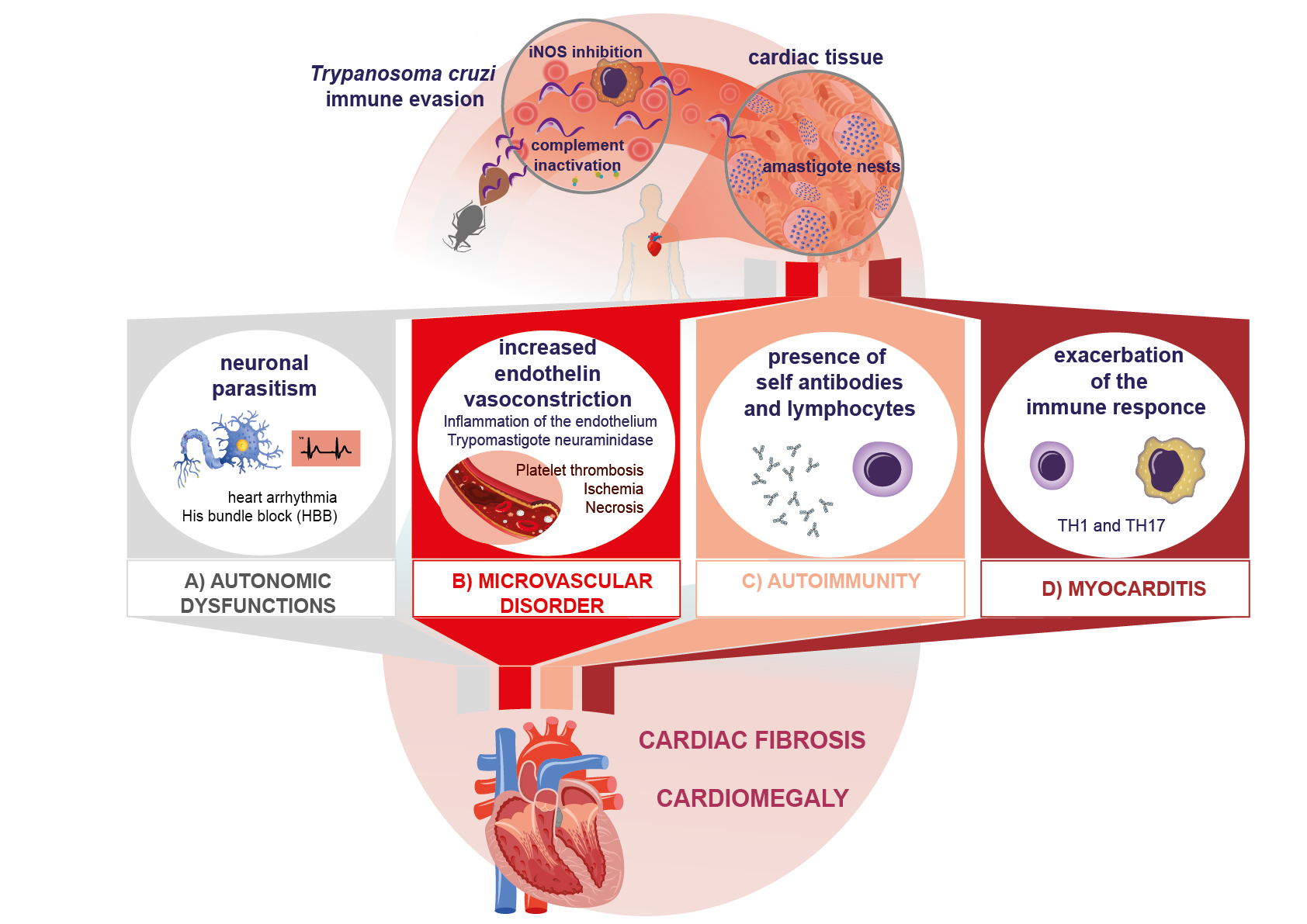
4. Autoimmune Mechanisms in Chagas Disease
5. Inflammatory and Cytotoxic Process in the Immunopathogenesis of Chagas Disease
6. Fibrotic Mechanisms in Chagas Disease
7. Conclusions
References
- Chagas, C. Nova Tripanozomiaze Humana: Estudos Sobre a Morfolojia e o Ciclo Evolutivo Do Schizotrypanum Cruzi n. Gen., n. Sp., Ajente Etiolojico de Nova Entidade Morbida Do Homem. Mem. Inst. Oswaldo Cruz 1909, 1, 159–218.
- World Health Organization. Asamblea Mundial de la Salud. In Enfermedad de Chagas: Control y eliminación; World Health Organization: Geneva, Swithzerand, 2009; p. 4.
- Organización Panamericana de la Salud. Enfermedad de Chagas. Available online: https://bit.ly/3HrzTwF (accessed on 6 January 2023).
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas Disease: From Discovery to a Worldwide Health Problem. Front. Public Health 2019, 7, 166.
- Secretaría de Salud Manual De Procedimientos Para La Enfermedad De Chagas En México; Secretaría de Salud: Ciudad de México, México, 2019; pp. 32–33.
- Salazar-Schettino, P.M.; Bucio Torres, M.I.; Cabrera-Bravo, M.; De Alba-Alvarado, M.; Castillo-Saldaña, D.R.; Zenteno-Galindo, E.; Rojo-Medina, J.; Fernández-Santos, N.; Perera-Salazar, M.G. Enfermedad de Chagas en México. Rev. Fac. Med. UNAM 2016, 59, 6–16.
- Salazar-Schettino, P.M.; Majumder, S.; Kierszenbaum, F. Regulatory Effect of the Level of Free Ca2+ of the Host Cell on the Capacity of Trypanosoma Cruzi to Invade and Multiply Intracellularly. J. Parasitol. 1995, 81, 597–602.
- Giordanengo, L.; Guiñazú, N.; Stempin, C.; Fretes, R.; Cerbán, F.; Gea, S. Cruzipain, a MajorTrypanosoma Cruziantigen, Conditions the Host Immune Response in Favor of Parasite. Eur. J. Immunol. 2002, 32, 1003–1011.
- Woolsey, A.M.; Sunwoo, L.; Petersen, C.A.; Brachmann, S.M.; Cantley, L.C.; Burleigh, B.A. Novel PI 3-Kinase-Dependent Mechanisms of Trypanosome Invasion and Vacuole Maturation. J. Cell Sci. 2003, 116, 3611–3622.
- Salassa, B.N.; Cueto, J.A.; Gambarte Tudela, J.; Romano, P.S. Endocytic Rabs Are Recruited to the Trypanosoma Cruzi Parasitophorous Vacuole and Contribute to the Process of Infection in Non-Professional Phagocytic Cells. Front. Cell. Infect. Microbiol. 2020, 10, 536985.
- Teixeira, A.R.L.; Hecht, M.M.; Guimaro, M.C.; Sousa, A.O.; Nitz, N. Pathogenesis of Chagas’ Disease: Parasite Persistence and Autoimmunity. Clin. Microbiol. Rev. 2011, 24, 592–630.
- De Diego, J.; Punzón, C.; Duarte, M.; Fresno, M. Alteration of Macrophage Function by a Trypanosoma Cruzi Membrane Mucin. J. Immunol. 1997, 159, 4983–4989.
- De Lima Rivero, A.R.; Farías Tamoy, M.N.; Tortolero Leal, E.; Navarro Aguilera, M.C.; Contreras Alvarez, V.T. . Acta Cient. Venez. 2001, 52, 235–247.
- Cummings, R.D.; Hokke, C.H.; Haslam, S.M. Parasitic Infections. In Essentials of Glycobiology, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; ISBN 978-1-62182-421-3.
- Nardy, A.F.F.R.; Freire-de-Lima, C.G.; Pérez, A.R.; Morrot, A. Role of Trypanosoma Cruzi Trans-Sialidase on the Escape from Host Immune Surveillance. Front. Microbiol. 2016, 7, 348.
- Cestari, I.; Ramirez, M.I. Inefficient Complement System Clearance of Trypanosoma Cruzi Metacyclic Trypomastigotes Enables Resistant Strains to Invade Eukaryotic Cells. PLoS ONE 2010, 5, e9721.
- Ferreira, V.; Valck, C.; Sánchez, G.; Gingras, A.; Tzima, S.; Molina, M.C.; Sim, R.; Schwaeble, W.; Ferreira, A. The Classical Activation Pathway of the Human Complement System Is Specifically Inhibited by Calreticulin from Trypanosoma Cruzi. J. Immunol. 2004, 172, 3042–3050.
- Doyle, P.S.; Zhou, Y.M.; Hsieh, I.; Greenbaum, D.C.; McKerrow, J.H.; Engel, J.C. The Trypanosoma Cruzi Protease Cruzain Mediates Immune Evasion. PLoS Pathog. 2011, 7, e1002139.
- Teixeira, M.M.; Gazzinelli, R.T.; Silva, J.S. Chemokines, Inflammation and Trypanosoma Cruzi Infection. Trends Parasitol. 2002, 18, 262–265.
- De Alba-Alvarado, M.; Bucio-Torres, M.I.; Zenteno, E.; Sampedro-Carrillo, E.; Hernández-Lopez, M.; Reynoso-Ducoing, O.; Torres-Gutiérrez, E.; Guevara-Gomez, Y.; Guerrero-Alquicira, R.; Cabrera-Bravo, M.; et al. Response to Infection by Trypanosoma Cruzi in a Murine Model. Front. Vet. Sci. 2020, 7, 568745.
- Jones, E.M.; Colley, D.G.; Tostes, S.; Lopes, E.R.; Vnencak-Jones, C.L.; McCurley, T.L. A Trypanosoma Cruzi DNA Sequence Amplified from Inflammatory Lesions in Human Chagasic Cardiomyopathy. Trans. Assoc. Am. Physicians 1992, 105, 182–189.
- Köberle, F. Chagas’ Disease and Chagas’ Syndromes: The Pathology of American Trypanosomiasis. In Advances in Parasitology; Elsevier: Amsterdam, Netherlands, 1968; Volume 6, pp. 63–116. ISBN 978-0-12-031706-6.
- Marin-Neto, J.A.; Cunha-Neto, E.; Maciel, B.C.; Simões, M.V. Pathogenesis of Chronic Chagas Heart Disease. Circulation 2007, 115, 1109–1123.
- Herrera, R.N.; Díaz, E.; Pérez-Aguilar, R.; Bianchi, J.; Berman, S.; Luciardi, H.L. Prothrombotic state in early stages of chronic Chagas’ disease. Its association with thrombotic risk factors. Arch. Cardiol. Méx. 2005, 75, 38–48.
- Teixeira, A.R.; Nascimento, R.J.; Sturm, N.R. Evolution and Pathology in Chagas Disease: A Review. Mem. Inst. Oswaldo Cruz 2006, 101, 463–491.
- Gutierrez, F.R.S.; Guedes, P.M.M.; Gazzinelli, R.T.; Silva, J.S. The Role of Parasite Persistence in Pathogenesis of Chagas Heart Disease: Parasite Persistence in Chagas Heart Disease. Parasite Immunol. 2009, 31, 673–685.
- Kierszenbaum, F. Views on the Autoimmunity Hypothesis for Chagas Disease Pathogenesis. FEMS Immunol. Med. Microbiol. 2003, 37, 1–11.
- Cunha-Neto, E.; Teixeira, P.C.; Nogueira, L.G.; Kalil, J. Autoimmunity. In Advances in Parasitology; Elsevier: Amsterdam, Netherlands, 2011; Volume 76, pp. 129–152. ISBN 978-0-12-385895-5.
- De Bona, E.; Lidani, K.C.F.; Bavia, L.; Omidian, Z.; Gremski, L.H.; Sandri, T.L.; Messias Reason, I.J. de Autoimmunity in Chronic Chagas Disease: A Road of Multiple Pathways to Cardiomyopathy? Front. Immunol. 2018, 9, 1842.
- Gironès, N.; Rodríguez, C.I.; Carrasco-Marín, E.; Hernáez, R.F.; de Rego, J.L.; Fresno, M. Dominant T- and B-Cell Epitopes in an Autoantigen Linked to Chagas’ Disease. J. Clin. Invest. 2001, 107, 985–993.
- Camargo, M.M.; Andrade, A.C.; Almeida, I.C.; Travassos, L.R.; Gazzinelli, R.T. Glycoconjugates Isolated from Trypanosoma Cruzi but Not from Leishmania Species Membranes Trigger Nitric Oxide Synthesis as Well as Microbicidal Activity in IFN-Gamma-Primed Macrophages. J. Immunol. 1997, 159, 6131–6139.
- Verdot, L.; Lalmanach, G.; Vercruysse, V.; Hartmann, S.; Lucius, R.; Hoebeke, J.; Gauthier, F.; Vray, B. Cystatins Up-Regulate Nitric Oxide Release from Interferon-γ- Activated Mouse Peritoneal Macrophages. J. Biol. Chem. 1996, 271, 28077–28081.
- Cardoni, R.L.; Antúnez, M.I.; Abrami, A.A. TH1 response in the experimental infection with Trypanosoma cruzi. Medicina 1999, 59, 84–90.
- Guiñazú, N.; Pellegrini, A.; Carrera-Silva, E.A.; Aoki, M.P.; Cabanillas, A.M.; Gìronés, N.; Fresno, M.; Cano, R.; Gea, S. Immunisation with a Major Trypanosoma Cruzi Antigen Promotes Pro-Inflammatory Cytokines, Nitric Oxide Production and Increases TLR2 Expression. Int. J. Parasitol. 2007, 37, 1243–1254.
- de Oliveira, T.B.; Pedrosa, R.C.; Filho, D.W. Oxidative Stress in Chronic Cardiopathy Associated with Chagas Disease. Int. J. Cardiol. 2007, 116, 357–363.
- Gupta, S.; Wen, J.-J.; Garg, N.J. Oxidative Stress in Chagas Disease. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 190354.
- Marchant, D.J.; Boyd, J.H.; Lin, D.C.; Granville, D.J.; Garmaroudi, F.S.; McManus, B.M. Inflammation in Myocardial Diseases. Circ. Res. 2012, 110, 126–144.
- Carvalho, C.M.E.; Silverio, J.C.; da Silva, A.A.; Pereira, I.R.; Coelho, J.M.C.; Britto, C.C.; Moreira, O.C.; Marchevsky, R.S.; Xavier, S.S.; Gazzinelli, R.T.; et al. Inducible Nitric Oxide Synthase in Heart Tissue and Nitric Oxide in Serum of Trypanosoma Cruzi-Infected Rhesus Monkeys: Association with Heart Injury. PLoS Negl. Trop. Dis. 2012, 6, e1644.
- Marino, A.P.M.P.; Silva, A.A.; Pinho, R.T.; Lannes-Vieira, J. Trypanosoma Cruzi Infection: A Continuous Invader-Host Cell Cross Talk with Participation of Extracellular Matrix and Adhesion and Chemoattractant Molecules. Braz. J. Med. Biol. Res. 2003, 36, 1121–1133.
- Talvani, A.; Ribeiro, C.S.; Aliberti, J.C.S.; Michailowsky, V.; Santos, P.V.A.; Murta, S.M.F.; Romanha, A.J.; Almeida, I.C.; Farber, J.; Lannes-Vieira, J.; et al. Kinetics of Cytokine Gene Expression in Experimental Chagasic Cardiomyopathy: Tissue Parasitism and Endogenous IFN-γ as Important Determinants of Chemokine MRNA Expression during Infection with Trypanosoma Cruzi. Microbes Infect. 2000, 2, 851–866.
- Machado, F.S.; Martins, G.A.; Aliberti, J.C.S.; Mestriner, F.L.A.C.; Cunha, F.Q.; Silva, J.S. Trypanosoma Cruzi—Infected Cardiomyocytes Produce Chemokines and Cytokines That Trigger Potent Nitric Oxide–Dependent Trypanocidal Activity. Circulation 2000, 102, 3003–3008.
- Araújo-Jorge, T.C.; Waghabi, M.C.; Soeiro, M.; Keramidas, M.; Bailly, S.; Feige, J.-J. Pivotal Role for TGF-β in Infectious Heart Disease: The Case of Trypanosoma Cruzi Infection and Consequent Chagasic Myocardiopathy. Cytokine Growth Factor Rev. 2008, 19, 405–413.
- Abel, L.C.J.; Rizzo, L.V.; Ianni, B.; Albuquerque, F.; Bacal, F.; Carrara, D.; Bocchi, E.A.; Teixeira, H.C.; Mady, C.; Kalil, J.; et al. Chronic Chagas’ Disease Cardiomyopathy Patients Display an Increased IFN-γ Response to Trypanosoma Cruzi Infection. J. Autoimmun. 2001, 17, 99–107.

