Chagas disease is a complex zoonosis. Its natural history involves the interaction of transmitting arthropods with wild, peridomestic, and domestic mammals, and it has a great diversity of transmission forms. In a vertebrate host, the disease has two clinical phases: an acute phase and a chronic one; the former evolves without demonstrated pathology and can last 10–20 years. After this phase, some patients progress to the chronic symptomatic phase, in which they develop mainly cardiac lesions. The lesions in this cardiomyopathy involve several cardiac tissues, mainly the myocardium, and in severe cases, the endocardium pericardium; this can cause pleural effusion, which may evolve into sudden death, which is more frequent in cases with dilated heart disease and severe heart failure.
1. Introduction
Chagas disease is a complex zoonosis. Its natural history involves the interaction of transmitting arthropods with wild, peridomestic, and domestic mammals, and it has a great diversity of transmission forms. In a vertebrate host, the disease evolution is evinced by various clinical manifestations [
1,
2].
The disease is caused by
Trypanosoma cruzi, a flagellated protozoan that is naturally transmitted by hematophagous Hemiptera insects (triatomines). The parasite was discovered in 1909 by Dr. Carlos Chagas in Minas Gerais, Brazil. Dr. Chagas also described a large part of the biological cycle, linking the parasite to the transmitting triatomine (
Panstrongylus megistus). He was able to isolate the parasite and replicated the infection in experimental animals. Dr. Chagas also mentioned that rural housing conditions, which to date have not changed significantly in endemic countries, are important for the spread of the vector. As a clinical entity, Chagas disease is linked to poverty [
1].
Chagas disease is endemic in 21 countries within continental Latin America. It is distributed from the south of the United States, in Central America, the Southern Cone, Andean countries, and Amazonian countries. In the Americas, 30,000 new cases are reported every year, 12,000 deaths on average, and 8600 newborns are infected at gestation. In 2019, a prevalence rate of 933.76 per 100,000 population was recorded. Currently, about 70 million people in the Americas live in areas at risk for the infection [
3]. Humans become infected with
T. cruzi by several mechanisms. The most important one is natural transmission, involving an infected triatomine bug.
4].CTh
is tra
gas disease has two clinical phases: an acute phase and a chronic one. Most cases of acute Chagas disease are asymptomatic; only 5–10% of infected subjects develop symptoms, including persistent fever, asthenia, adynamia, headache, and hepatosple-nomegaly, all of which are nonspecific. Among these symptomatic individuals,nsmission form is very common in rural areas, where housing traits and the ecotope favor a colonization of the domestic niche by insects. The second transmission form, restricted to urban areas, is related to the transfusion of blood or its components. This transmission form depends on the m
ost frequent pathognomonic signs are the Romaña sign, characterized by unilateral bi-palpebral edema, wigration of rural population to cities, as more than 70% of the population in some cities had immigrated from high-prevalence areas of the disease [4].
The prich is observedmary vectors for Chagas Disease are Triatoma infestans in 50%Argentina, of cases, and the indurated cutaneous chagoma, found Bolivia, Brazil, Chile, Paraguay, Uruguay, and Peru; Rhodnius prolixus in 25% Colof cases.
Bmbia, Vene
nz
nidazole and nifurtimox areuela, and Central America; theT. dimidiata oin
ly drugs with proven efficacy aga Ecuador and Central America; and R. pallescens in
st Chagas diseasePanama [3].
BoIn th
drugs have been approved internationally. Antiparasitic treatment of these cardiomyopathies is accompanied by the administre Americas, natural infection is associated with risk factors such as housing construction material and other characteristics that favor the colonization of
antiarrhythmics and pace-maker placement. In the most severe cases, heart transplantation can be required [5,7].
2. Pathogenic Mechanisms of Immune Response Evasion in Chagas Disease
Mohuman dwellings in rural areas. For this reason, it is considered as
t of the mechanisms involveda neglected tropical disease [4]. inPAHO/WHO, evading the immune response occur in the acute phase, when trypomastigotes establish contact with immune cells of the vertebrate host. The parasite has evolved mechanisms to survive processes such as phagocytosis and the complement systemworking with the endemic countries, have launched several Subregional Disease Prevention and Control Initiatives. These include the improvement of housing to halt vectorial transmission in 17 countries, and screening of blood donors in the 21 endemic countries, in addition to
interfering with lymphocyte maturateliminating some vector species such as R. prolixus i
on
. When metacyclic trypomastigotes contact host cells, e El Salvador, Costa Rica, and Mexico, and T. infestans i
then Br
througazil and Uruguay [3,5].
Due to th
e skin lesions or mucous membranes, the immune response is activated.increase in population mobility worldwide, Chagas disease is considered T. cruzia entemajor
s cells through two main mechanisms. The first one is lysosome-dependent, which favors Ca2+ health problem, which has reached countries where vector transmission does not exist, due to the im
obilizmigration
[18].of In this
stage, the parasite surface glycoprotein gp82 is crucial for cell adhesion and lysosomal fusion at the site of entry. Cruzipain has also been proven to be critical for calcium induction and lysosome recruitment. It is a cysteine-protease secreted by trypomastigotes [19,20]. Teropositive cases from endemic geographic areas. This poses a risk of transmission through transfusions, organ and tissue transplants, and even maternal-fetal transmission in the USA, Canada, and some European and Western Pacific countries. It has been estimated that Spain is the
second m
echanism involves invagination of the plasma membraneain non-endemic country in the number of transmissions, followed by
ly the USA and Italy [6].
Chagas
o disease has
omal fusion. The acidification process of lysosomes containing the parasite is key to its differentiation into an amastigote, which is the replicative form. A disruption of cytoskeletal actin facilitates two clinical phases: an acute phase and a chronic one. Most cases of acute Chagas disease are asymptomatic; only 5–10% of infected subjects develop symptoms, including persistent fever, asthenia, adynamia, headache, and hepatosple-nomegaly, all of which are nonspecific. Among these symptomatic individuals, the mo
bilization of lysosomes towards the cell periphery, where they will fuse with the cytoplasmic membrane and contribute to the formation of a parasitic vacuole [21]st frequent pathognomonic signs are the Romaña sign, characterized by unilateral bi-palpebral edema, which is observed in 50% of cases, and the indurated cutaneous chagoma, found in 25% of cases.
AfBot
er several divisions in the parasitophorous vacuole, where sialic acid residues are added oh signs are often accompanied by regional adeno-megaly. In the
T. cruzi mrem
bra
ne by parasitic trans-sialidases, the phagolysosome is lysed. Thus, trypomastigotes are released intoining 25% of patients, there is no sign of portal entry, but some of the nonspecific symptoms mentioned above can be found [7]. tThe
cytoplasm and bloodstream to infect distant or adjacent tissues and cells [22]. Notmost frequent symptom is fever, which is present in up to 95% of cases, usua
blly
, factors such as strain, the level of antioxidant enzyme expression without specific characteristics. All other signs and symptoms, including asthenia, adynamia, headache, and
the kineticshepatosple-nomegaly, are nonspecific [3,5]. While ofT. cruzi assoc
iation with the phagolysosome may in turn contribute to parasite evasion and persistence in the host. In contrast toan infect any nucleated cell, some strains exhibit a marked tropism for myocardial cells, smooth muscle cells of the digestive system, or nervous tissue, among other
protozoa, which inhibit phagolysosome maturation,cell types. Cardiac manifestations include primarily organ T. cruzi e
vnla
de margement (cardiomegaly) [7].
The c
hro
phage activity by the mechanisms mentioned above, and escape from the phagolysosome into the host cell cytoplasm, where they replicate [29].
Thenic phase is divided into a chronic asymptomatic phase and a symptomatic phase; the former evolves without demonstrated pathology and can last 10–20 years; however, cases have been reported in cmino
mplement system features a specialized pathway for mannose recognition in pathogenic organisms, and it is known that blood trypomastigotes activate rs in Mexico where this period lasts 2–7 years before the chronic form with cardiovascular symptomatology can be detected [8]; this
phas
ystem; however, thee is clinically asymptomatic and exhibits very low parasite
expresses a set of specific surface proteins for complement evasion [30,31]. T. cruzi tramia, so that the methods of choice for diagnosis are serological, and con
s-sfi
alidases are crucial for host cell infection by transrmation requires two positive tests with different principles [9].
Af
ter
ring sialic acid from mammalian cell this phase, patients progress to the
ir own glycocalyx [32]. T chronic symptomatic ph
ase
ir presence is also known to reduce the recognition of anti-α-Gal antibodies in the bloodstream, in addition to evading the lytic effect of complement by promoting the conversion of C3 to inactive C3b (iC3b) [33].
T. cruzi ca, with a proven symptomatology, in which they develop mainly cardiac lesions and, to a lesser extent, digestive lesions, mainly in the esophagus and colon, and in a few cases in the peripheral nervous system. Cardiac lesions cause alterations in myocardial contractility and the el
re
ticulin (TcCRT) is also involvedctrical impulse, mainly in the
evasion of the lectin pathway by binding to MBL or inhibiting the classical complement pathway by binding to C1qbundle of His, the particular right bundle branch block with left anterior fascicular hemiblock, ventricular extrasystoles, and atrioventricular block [3,7]. The
se l
inks inactivate the membrane attack complex (MAC) formation pathway [34]. Aesions in this cardiomyopathy involve several cardiac tissues, mainly the myocardium, and in severe cases, the en
do
ther mechanism by which the parasite controls the complement pathway is mediated by the cardium pericardium; this can cause pleural effusion, which may evolve into sudden death, which is more frequent in cases with dilated heart disease and severe heart failure [10]. C
RP par
otein, which is anchorlos Chagas described digestive disorders linked to the
parasite membrane. This 160-kDa glycoprotein binds non-covalently to C3b and C4b to inhibit the assembly of C3 convertase, rendering it inactive to catalyze the cleavagedisease in 1916, although Kidder and Fletcher had already done so in 1857, when they called it mal d’engasgo, i.e., “disease causing dysphagia” [11]. Eso
f the complphageal involvement
system on the parasite surface. Another parasitic protein called T-DAF accelerates the decay of C3 and C5 convertase in the classical and alternative pathways of the complement system.usually consists of a megaesophagus with slow esophageal transit disorders, along with pain and difficulty in swallowing. In cases of colon involvement, a megacolon and constipation are typical [
3512,13].
3. Histopathological Mechanisms Related to Tissue Parasitism
T Chagas he
art presence and replication by binary fission of intracellular amastigotes in the mydisease is clinically classified according to symptoms, and electrocardio-graphic, echocardio
cyte and its ensuing lysis cause inflammation, the release of cellular components, and finally the destruction of cardiac tissue [36].graphic, and radiological abnormalities, especially changes in left ventricular function. Some risk factors predisposing one to Aa direct correlation between the presence of the parasite and tissue inflammation has been reported [37]progression to the chronic phase include electrocardiographic abnormalities,
male
ven when only the presence of T. cruzi sex, systolic blood pressure less than
tigens or DNA has been confirmed in chronic lesions [38]. The infe120 mmHg, altered systolic function
, can affect skeletal, cardiacleft ventricular dilatation, and
smooth muscle, as well as neuronal tissue. Parasitism in the myocardium causes destrucomplex arrhythmias; risk scores have been proposed to stratify the risk of death, although their clinical value is still under study [14]. In a c
tion
and fibrosis in the Schwann sheath and nerve fibers, which leads to neurogenic alterations, clinically expressed as arrhythmias, His bundle branch block, and even the presence of dyssynergic areas in the parietal funcsensus, several authors note that the main predictors of poor prognosis in chronic Chagas disease are a deterioration of left ventricular function, falling into classes III (fatigue, palpitations, dyspnea, or anginal pain) and VI (cardiomegaly and non-sustained ventricular tachycardia) of the New York Heart Association (NYHA) classification of
the heart (Figure 1A). Aheart fail
lure of[15]. Othe
se are characteristic manifestations of the chronic phase of Chagas diseaser risk scores have been proposed. Rassi uses a combination of clinical symptoms, cabinet test results, and demographic data [
39,4016].
AOn
other important mechanism described in myocardial injury is the occurrence of microvas the other hand, de Sousa use a four-factor score that includes the QT dispersion interval on ECG, syncope, premature ventricular
abnormalities caused by the increase in contractions, and left ventricular function [17].
Ben
znid
othelin due to inflammation. Onazole and nifurtimox are the o
ther hand, blood trypomastigotenly drugs with proven efficacy against Chagas disease. Both drugs have been
described to produce neuraminidase (Figure 1B)approved internationally. A
ll nt
hese mechanisms increase platelet adhesiveness and thrombus formation, leading to myocardial ischemia and microinfarcts, which in turn cause tissue necrosisiparasitic treatment of these cardiomyopathies is accompanied by the administration of antiarrhythmics and pace-maker placement. In the most severe cases, heart transplantation can be required [
415,
427].
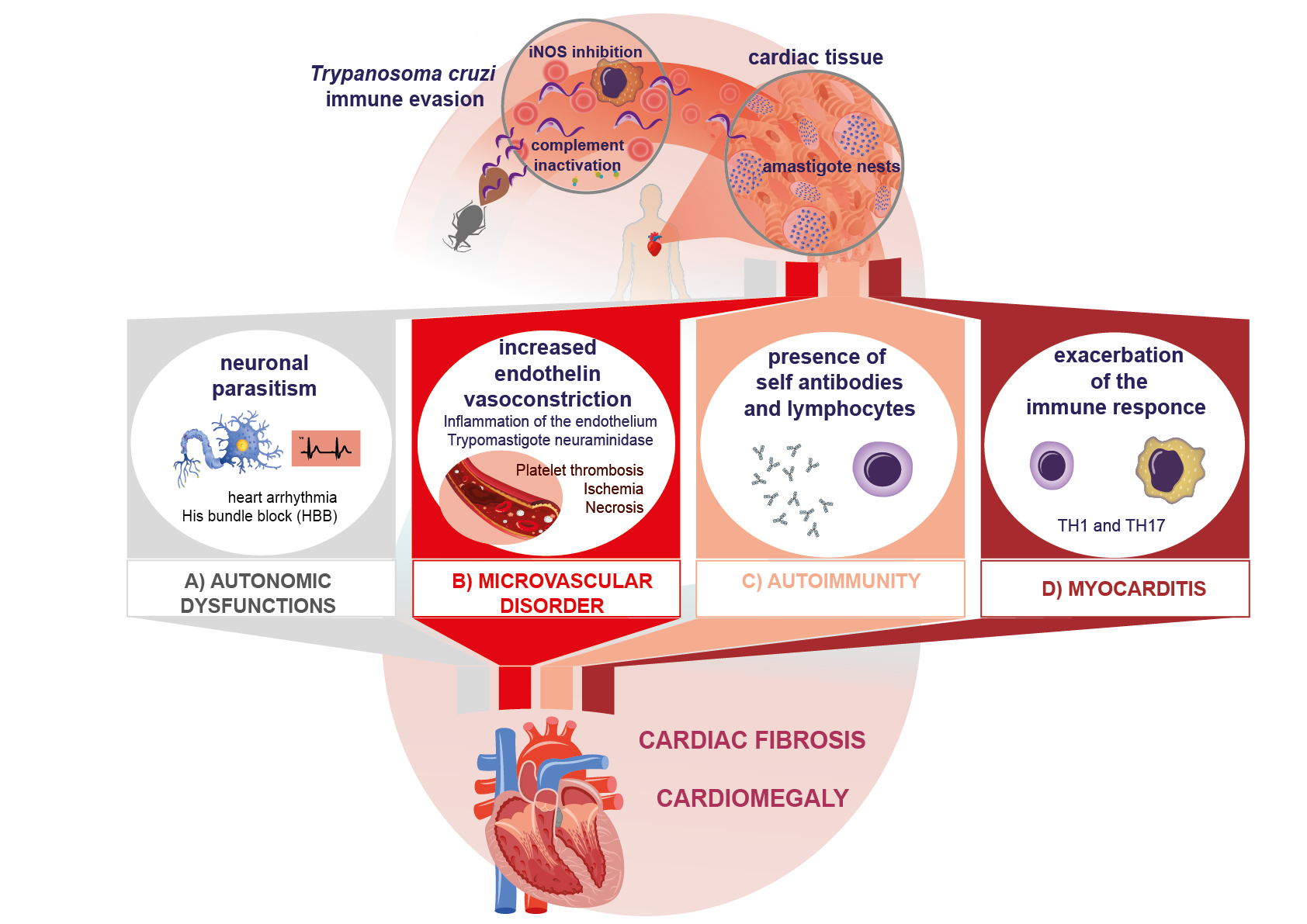
42. AuPatoimmunehogenic Mechanisms in of Immune Response Evasion in Chagas Disease
SeveralMost of the
ories have been advanced on the development of lesions in heart disease. Among them, the presence of auto mechanisms involved in evading the immune response occur in the acute phase, when trypomastigotes establish contact with immune
phenomena and the persistence of tcells of the vertebrate host. The parasite
stand out, without being mutually exclusive [47]. Thas evolved mechanisms to survive pro
datcesse
, there is no consensus regarding the autoimmune hypothesis in the pathogenesis of Chagas disease; however, several studies have demonstrated the participation of mechanisms that could contribute to tissue damage in the chronic phase [47s such as phagocytosis and the complement system, in addition to interfering with lymphocyte maturation. When metacyclic trypomastigotes contact host cells, either through skin lesions or mucous membranes,
48]. Aut
ohe immune
damage processes canresponse is activated. T. cruzi origien
ate ters cells through
severaltwo main mechanisms
: (1) In the acute phase, lysis of infected vertebrate cells leads to . The first one is lysosome-dependent, which favors Ca
n2+ expmo
sure of cryptic antigensbilization [18]. orIn with molecular mimicry betweenthis stage, the parasite
and host cell epitopes. (2) The release and presentation of self-antigens. The ensuing production of inflammatory cytokines may exceed the activation threshold, breaking self-tolerance and stimulating T and B lymphocytes [49]. (3) T csurface glycoprotein gp82 is crucial for cell adhesion and lysosomal fusion at the site of entry. Cruzipain has also been proven to be critical for calcium induction and lysosome re
ll ac
tivation without cytokine signaling (bystander activation). (4) Autoantibody-dependent cell-mediated cytotoxicityruitment. It is a cysteine-protease secreted by trypomastigotes [
5019,20].
AllThe foursecond mechanism
s may result in autoimmune damage.
Oth involves invagination of the plasma membrane
r processes inherent to the presentation of host antigens after myocardial lysis suggest the existence of cryptic antigens. This is the case in traumatic ophthalmopathiesfollowed by lysosomal fusion. The acidification process of lysosomes containing the parasite is key to its differentiation into an amastigote, which
expose previously cryptic antigens. Since tolerance for these antigens has not been developed, autoreactive clones are produced. Similarly, some cryptic epitopes can show molecular mimicry with T. cruzi epis the replicative form. A disruption of cytoskeletal actin facilitates the mobilization of lysosomes towards the cell periphery, where they will fuse with the cytoplasmic membrane and contri
but
opes, which favors the genere to the formation of
these clonesa parasitic vacuole [
6421].
Such a pAfter
ocess has been reported in Chagas disease after host cell lysis (Figure 1C).
5. Inflammatory and Cytotoxic Process in the Immunopathogenesis of Chagas Disease
T several divisions in the parasitophorous vacuole, where sialic acid residues are added on the
inflaT. cruzi m
em
atory process in Chagas disease starts in the acute phase, with the production of proinflammatory cytokines to recruit and induce thebrane by parasitic trans-sialidases, the phagolysosome is lysed. Thus, trypomastigotes are released into the cytoplasm and bloodstream to infect distant or adjacent tissues and cells [22].
T. cruzi ac
tivation of monocytes toan also be phagocytosed at the site of infection
. This proces by tissue macrophages. Leishmania s
pp. is important to control parasite replication throughalso parasitizes macrophages and develops mechanisms similar to those of T. cruzi a[23]. Th1-typSome
reLeishmania sp
onecies
e, characterized by the production of interleukins (IL) such as that parasitize cutaneous and peripheral blood macrophages and have been isolated in Latin America are L. mexicana, L. amazonensis, IL-1L. venezuelensis,
and L. braziliensis; in
t
er-feron gammahe Mediterranean, we find (IFN-γ)L. infantum, and
in Asia th
e tumorere are L. tropica an
ecrosisd L. major [23]. facIt
or alpha (TNF-α). The functions of IFN-γ predomin should be emphasized that these species, phylogenetically related to T. cruzi a
tes in this mechanism by inducing the produKinetoplastida, can inhibit the antiparasitic function of
nitric oxide (NO) in macrophages
and initiating the control of blood parasitemia, and both genera use this strategy; in th
is phe case of
Leishmania spp., the
y disease [74]. Ocreate a safe in
t
he other hand, it has also been reported that IL-10 and IL-4, stimulated by antigens such asracellular compartment and continue their life cycle in the mammal, and in the case of cruzipainT. cruzi,
rthey e
ducvade the p
roduction of IFN-γ and NO in macrophages, inhibiting phagocytosishagolysosome and escape to the cytosol for replication [
7524,25].
As nTo
ted, while the Th1 response protects the host in the acute phase, its survive in this extremely oxidative environment inside the macrophages, the T.cruzi de
regulation in the chronic phase leads to cardiac tissue damage by exacerbated, chronic inflammation (Figure 1D) [76]. Molxpress antioxidant enzymes such as peroxidases, which protect it from reactive oxygen and nitrogen spec
ulies
that contribute towithin macrophages [26,27]. In t
his
sue damage (DAMPS), specific regard, an overexpression of TcCPX in T. cruzi ha
llys been of cytotoxic origin, are generated by shown to correlate with increased parasitemia and inflammat
ionory infiltrates in the myocardium [
7728].
Notably, Infactors this regard, there is evidence that oxidative stress also induces myocardial lesions in these patients,such as strain, the level of antioxidant enzyme expression, and the kinetics of association with the phagolysosome may in turn contribut
ing to lesion progression in Chagas disease [78].
Ase to parasite evasion and persistence in the host. In contrast to othe
condition progresses towr protozoa, which inhibit phagolysosome maturation, T. cruzi eva
rd
s chronicity, furthere macrophage activity by the mechanisms
of cytotoxic damage affect cardiac tissue. Initially, oxidative stress is triggered, and reactive oxygementioned above, and escape from the phagolysosome into the host cell cytoplasm, where they replicate [29]. On
spec
ies are released. These reactive oxygen species, mainly H2O2 ae outside the macrophages or in any in
fected
OHtissue,
T. cruzi can be
pr
o-ducecognized by
cytotoxic immune responses that cause lysis in the tissues surrounding the infec-tion [79,80]their different PAMPs, which are mainly glycoinositolphospholipids (GIPLs) and lipopepti-doglycans (LPPG).
AnotThe
r mechanism triggered by the production of reactive oxygen species is the increase in NO levels, which stimulates the production of proinflammatory cytokinese molecules have protective functions because they allow the parasite to survive in hydrolytic environments and
the activation of adhesion molecules for rolling, as well as an increased endothelial permeability [81].
6. Fibrotic Mechanisms in Chagas Disease
Cardiac tipromote adherence to mammalian cells for invasion. The complement s
ys
ue fibrosis is accompanied by cellular inflammatory infiltrates characterized by the presence of macrophages, neutrophiltem features a specialized pathway for mannose recognition in pathogenic organisms, and
eosinophils during the acute phase, and by lymphocytic infiltrates in the chronic phase. Acute or chronic fibrosis has been describedit is known that blood trypomastigotes activate this system; however, the parasite expresses a set of specific surface proteins for complement evasion [30,31]. inT. cruzi humtrans
by immunohistochemistry in postmortem studies, pointing to the relevance of these proinflammatory-sialidases are crucial for host cell infection by transferring sialic acid from mammalian cells
and fibrotic accompanimentto their own glycocalyx [
91,9232].
CaTheir pr
diomyocytes have also been shown to actively participateesence is also known to reduce the recognition of anti-α-Gal antibodies in the
inflammatory responsebloodstream, in addition to evading the lytic effect of complement by pr
oducing chemokines, cytokines, and NO. These factors can triggeromoting the conversion of C3 to inactive C3b (iC3b) [33].
T. cruzi cal
eukocytes at-traction, and they have even been suggested to promote fibrosis by secreting fibro-blast-activating growth factors and cytokines [93]. Ireticulin (TcCRT) is also involved in the evasion of the lectin pathway by binding to MBL or inhibiting the classical complement pat
h
as been experimentally proven in primary cultures that soluble mediators synthesized by cardiomyocytes can regulate fibronectinway by binding to C1q. These links inactivate the membrane attack complex (MAC) formation pathway [34]. syAn
thesis [94].
Vother mecha
rni
ous works have studied in vitro the process of the extracellular matrix remodeling due to the presence of cytokines, mainly TGF-β, given that fibrosis is a characteristic process in Chagas chronicity. Other studies have reported that both TGF-β and IL-10 are major regulators of the Th1 response in the lapse preceding the onset of tissue damage, and decreasing the activation of phagocytic cells. On the other hand, TGF-β, IL-13, and IFN-γ are directly related to the severity of cardiac dysfunction due to degenerative fibrosis, which induces motility disorders and organ growth [94,95].
7. Conclusions
Asm by which the parasite controls the complement pathway is mediated by the CRP protein, which is anchored to the parasite membrane. This 160-kDa glycoprotein binds non-covalently to C3b and C4b to inhibit the assembly of C3 convertase, rendering it inactive to catalyze the cleavage of the complement system on the parasite surface. Another parasitic protein called T-DAF accelerates the decay of C3 and C5 convertase in the classical and alt
oge
ther, the evasionrnative pathways of the
immune response bycomplement system. The mucin-rich surface of T. cruzi can
d the proinflammatory immune response lead to autonomic heart dysfunction and microvascular disorders. They also activate autoimmune processes that result in the fibrosis that characterize be recognized by Toll-like receptors, such as TLR-2, expressed on mac-rophages. This interaction induces the synthesis of proinflammatory cytokines such as IL-12 and TNF-α, and it favors the
chronic phase activation of the i
nfection. These mechanisms, triggeredNOS pathway in these phagocytes. In addition, cruzipain from byT. cruzi th
e presence of the parasite or its antigens, are responsible for the processes that underlie the immunopathogenesis of cardiac lesions in Chagas diseaseas a proteolytic action on the NF-κB protein complex, inhibiting the transcription of proinflammatory cytokines such as IL-12 [35].