Chagas disease is a complex zoonosis. Its natural history involves the interaction of transmitting arthropods with wild, peridomestic, and domestic mammals, and it has a great diversity of transmission forms. In a vertebrate host, the disease has two clinical phases: an acute phase and a chronic one; the former evolves without demonstrated pathology and can last 10–20 years. After this phase, some patients progress to the chronic symptomatic phase, in which they develop mainly cardiac lesions. The lesions in this cardiomyopathy involve several cardiac tissues, mainly the myocardium, and in severe cases, the endocardium pericardium; this can cause pleural effusion, which may evolve into sudden death, which is more frequent in cases with dilated heart disease and severe heart failure.
1. Introduction
Chagas disease is a complex zoonosis. Its natural history involves the interaction of transmitting arthropods with wild, peridomestic, and domestic mammals, and it has a great diversity of transmission forms. In a vertebrate host, the disease evolution is evinced by various clinical manifestations [
1,
2].
The disease is caused by
Trypanosoma cruzi, a flagellated protozoan that is naturally transmitted by hematophagous Hemiptera insects (triatomines). The parasite was discovered in 1909 by Dr. Carlos Chagas in Minas Gerais, Brazil. Dr. Chagas also described a large part of the biological cycle, linking the parasite to the transmitting triatomine (
Panstrongylus megistus). He was able to isolate the parasite and replicated the infection in experimental animals. Dr. Chagas also mentioned that rural housing conditions, which to date have not changed significantly in endemic countries, are important for the spread of the vector. As a clinical entity, Chagas disease is linked to poverty [
1].
Chagas disease is endemic in 21 countries within continental Latin America. It is distributed from the south of the United States, in Central America, the Southern Cone, Andean countries, and Amazonian countries. In the Americas, 30,000 new cases are reported every year, 12,000 deaths on average, and 8600 newborns are infected at gestation. In 2019, a prevalence rate of 933.76 per 100,000 population was recorded. Currently, about 70 million people in the Americas live in areas at risk for the infection [
3]. Humans become infected with
T. cruzi by several mechanisms. The most important one is natural transmission, involving an infected triatomine bug.
4].
Chagas disease has two clinical phases: an acute phase and a chronic one. Most cases of acute Chagas disease are asymptomatic; only 5–10% of infected subjects develop symptoms, including persistent fever, asthenia, adynamia, headache, and hepatosple-nomegaly, all of which are nonspecific. Among these symptomatic individuals, the most frequent pathognomonic signs are the Romaña sign, characterized by unilateral bi-palpebral edema, which is observed in 50% of cases, and the indurated cutaneous chagoma, found in 25% of cases.
Benznidazole and nifurtimox are the only drugs with proven efficacy against Chagas disease. Both drugs have been approved internationally. Antiparasitic treatment of these cardiomyopathies is accompanied by the administration of antiarrhythmics and pace-maker placement. In the most severe cases, heart transplantation can be required [
5,
7].
2. Pathogenic Mechanisms of Immune Response Evasion in Chagas Disease
Most of the mechanisms involved in evading the immune response occur in the acute phase, when trypomastigotes establish contact with immune cells of the vertebrate host. The parasite has evolved mechanisms to survive processes such as phagocytosis and the complement system, in addition to interfering with lymphocyte maturation. When metacyclic trypomastigotes contact host cells, either through skin lesions or mucous membranes, the immune response is activated.
T. cruzi enters cells through two main mechanisms. The first one is lysosome-dependent, which favors Ca
2+ mobilization [
18]. In this stage, the parasite surface glycoprotein gp82 is crucial for cell adhesion and lysosomal fusion at the site of entry. Cruzipain has also been proven to be critical for calcium induction and lysosome recruitment. It is a cysteine-protease secreted by trypomastigotes [
19,
20]. The second mechanism involves invagination of the plasma membrane followed by lysosomal fusion. The acidification process of lysosomes containing the parasite is key to its differentiation into an amastigote, which is the replicative form. A disruption of cytoskeletal actin facilitates the mobilization of lysosomes towards the cell periphery, where they will fuse with the cytoplasmic membrane and contribute to the formation of a parasitic vacuole [
21]. After several divisions in the parasitophorous vacuole, where sialic acid residues are added on the
T. cruzi membrane by parasitic trans-sialidases, the phagolysosome is lysed. Thus, trypomastigotes are released into the cytoplasm and bloodstream to infect distant or adjacent tissues and cells [
22]. Notably, factors such as strain, the level of antioxidant enzyme expression, and the kinetics of association with the phagolysosome may in turn contribute to parasite evasion and persistence in the host. In contrast to other protozoa, which inhibit phagolysosome maturation,
T. cruzi evade macrophage activity by the mechanisms mentioned above, and escape from the phagolysosome into the host cell cytoplasm, where they replicate [
29].
The complement system features a specialized pathway for mannose recognition in pathogenic organisms, and it is known that blood trypomastigotes activate this system; however, the parasite expresses a set of specific surface proteins for complement evasion [
30,
31].
T. cruzi trans-sialidases are crucial for host cell infection by transferring sialic acid from mammalian cells to their own glycocalyx [
32]. Their presence is also known to reduce the recognition of anti-α-Gal antibodies in the bloodstream, in addition to evading the lytic effect of complement by promoting the conversion of C3 to inactive C3b (iC3b) [
33].
T. cruzi calreticulin (TcCRT) is also involved in the evasion of the lectin pathway by binding to MBL or inhibiting the classical complement pathway by binding to C1q. These links inactivate the membrane attack complex (MAC) formation pathway [
34]. Another mechanism by which the parasite controls the complement pathway is mediated by the CRP protein, which is anchored to the parasite membrane. This 160-kDa glycoprotein binds non-covalently to C3b and C4b to inhibit the assembly of C3 convertase, rendering it inactive to catalyze the cleavage of the complement system on the parasite surface. Another parasitic protein called T-DAF accelerates the decay of C3 and C5 convertase in the classical and alternative pathways of the complement system. [
35].
3. Histopathological Mechanisms Related to Tissue Parasitism
The presence and replication by binary fission of intracellular amastigotes in the myocardiocyte and its ensuing lysis cause inflammation, the release of cellular components, and finally the destruction of cardiac tissue [
36]. A direct correlation between the presence of the parasite and tissue inflammation has been reported [
37], even when only the presence of
T. cruzi antigens or DNA has been confirmed in chronic lesions [
38]. The infection can affect skeletal, cardiac, and smooth muscle, as well as neuronal tissue. Parasitism in the myocardium causes destruction and fibrosis in the Schwann sheath and nerve fibers, which leads to neurogenic alterations, clinically expressed as arrhythmias, His bundle branch block, and even the presence of dyssynergic areas in the parietal function of the heart (
Figure 1A). All of these are characteristic manifestations of the chronic phase of Chagas disease [
39,
40]. Another important mechanism described in myocardial injury is the occurrence of microvascular abnormalities caused by the increase in endothelin due to inflammation. On the other hand, blood trypomastigotes have been described to produce neuraminidase (
Figure 1B). All these mechanisms increase platelet adhesiveness and thrombus formation, leading to myocardial ischemia and microinfarcts, which in turn cause tissue necrosis [
41,
42].
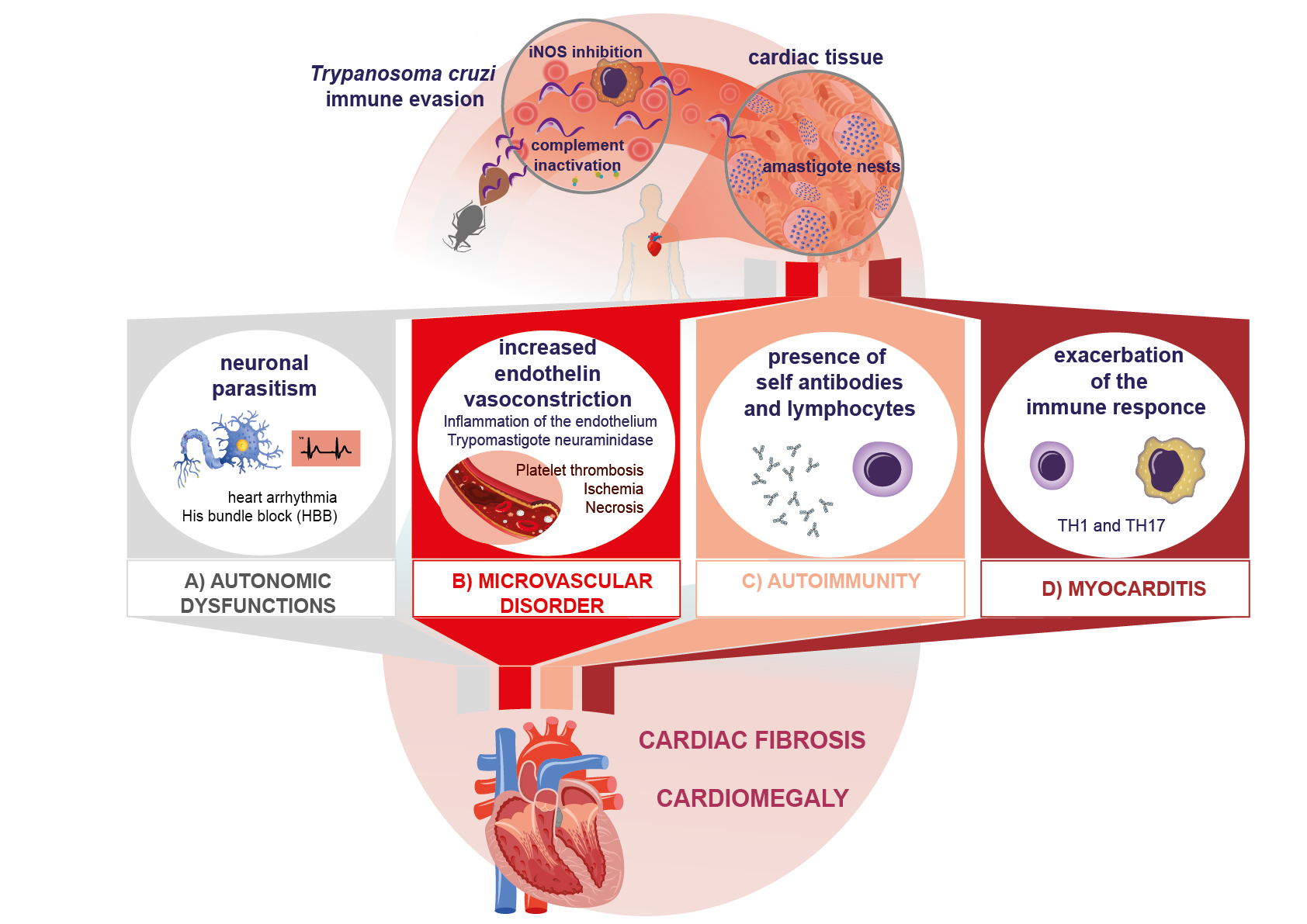
4. Autoimmune Mechanisms in Chagas Disease
Several theories have been advanced on the development of lesions in heart disease. Among them, the presence of autoimmune phenomena and the persistence of the parasite stand out, without being mutually exclusive [
47]. To date, there is no consensus regarding the autoimmune hypothesis in the pathogenesis of Chagas disease; however, several studies have demonstrated the participation of mechanisms that could contribute to tissue damage in the chronic phase [
47,
48]. Autoimmune damage processes can originate through several mechanisms: (1) In the acute phase, lysis of infected vertebrate cells leads to an exposure of cryptic antigens or with molecular mimicry between the parasite and host cell epitopes. (2) The release and presentation of self-antigens. The ensuing production of inflammatory cytokines may exceed the activation threshold, breaking self-tolerance and stimulating T and B lymphocytes [
49]. (3) T cell activation without cytokine signaling (bystander activation). (4) Autoantibody-dependent cell-mediated cytotoxicity [
50]. All four mechanisms may result in autoimmune damage.
Other processes inherent to the presentation of host antigens after myocardial lysis suggest the existence of cryptic antigens. This is the case in traumatic ophthalmopathies, which expose previously cryptic antigens. Since tolerance for these antigens has not been developed, autoreactive clones are produced. Similarly, some cryptic epitopes can show molecular mimicry with
T. cruzi epitopes, which favors the generation of these clones [
64]. Such a process has been reported in Chagas disease after host cell lysis (
Figure 1C).
5. Inflammatory and Cytotoxic Process in the Immunopathogenesis of Chagas Disease
The inflammatory process in Chagas disease starts in the acute phase, with the production of proinflammatory cytokines to recruit and induce the activation of monocytes to the site of infection. This process is important to control parasite replication through a Th1-type response, characterized by the production of interleukins (IL) such as IL-1, inter-feron gamma (IFN-γ), and the tumor necrosis factor alpha (TNF-α). The functions of IFN-γ predominate in this mechanism by inducing the production of nitric oxide (NO) in macrophages and initiating the control of blood parasitemia in this phase of the disease [
74]. On the other hand, it has also been reported that IL-10 and IL-4, stimulated by antigens such as cruzipain, reduce the production of IFN-γ and NO in macrophages, inhibiting phagocytosis [
75].
As noted, while the Th1 response protects the host in the acute phase, its deregulation in the chronic phase leads to cardiac tissue damage by exacerbated, chronic inflammation (
Figure 1D) [
76]. Molecules that contribute to tissue damage (DAMPS), specifically of cytotoxic origin, are generated by inflammation [
77]. In this regard, there is evidence that oxidative stress also induces myocardial lesions in these patients, contributing to lesion progression in Chagas disease [
78].
As the condition progresses towards chronicity, further mechanisms of cytotoxic damage affect cardiac tissue. Initially, oxidative stress is triggered, and reactive oxygen species are released. These reactive oxygen species, mainly H
2O
2 and OH, can be pro-duced by cytotoxic immune responses that cause lysis in the tissues surrounding the infec-tion [
79,
80]. Another mechanism triggered by the production of reactive oxygen species is the increase in NO levels, which stimulates the production of proinflammatory cytokines and the activation of adhesion molecules for rolling, as well as an increased endothelial permeability [
81].
6. Fibrotic Mechanisms in Chagas Disease
Cardiac tissue fibrosis is accompanied by cellular inflammatory infiltrates characterized by the presence of macrophages, neutrophils, and eosinophils during the acute phase, and by lymphocytic infiltrates in the chronic phase. Acute or chronic fibrosis has been described in humans by immunohistochemistry in postmortem studies, pointing to the relevance of these proinflammatory cells and fibrotic accompaniment [
91,
92]. Cardiomyocytes have also been shown to actively participate in the inflammatory response by producing chemokines, cytokines, and NO. These factors can trigger leukocytes at-traction, and they have even been suggested to promote fibrosis by secreting fibro-blast-activating growth factors and cytokines [
93]. It has been experimentally proven in primary cultures that soluble mediators synthesized by cardiomyocytes can regulate fibronectin synthesis [
94].
Various works have studied in vitro the process of the extracellular matrix remodeling due to the presence of cytokines, mainly TGF-β, given that fibrosis is a characteristic process in Chagas chronicity. Other studies have reported that both TGF-β and IL-10 are major regulators of the Th1 response in the lapse preceding the onset of tissue damage, and decreasing the activation of phagocytic cells. On the other hand, TGF-β, IL-13, and IFN-γ are directly related to the severity of cardiac dysfunction due to degenerative fibrosis, which induces motility disorders and organ growth [
94,
95].
7. Conclusions
Altogether, the evasion of the immune response by T. cruzi and the proinflammatory immune response lead to autonomic heart dysfunction and microvascular disorders. They also activate autoimmune processes that result in the fibrosis that characterizes the chronic phase of the infection. These mechanisms, triggered by the presence of the parasite or its antigens, are responsible for the processes that underlie the immunopathogenesis of cardiac lesions in Chagas disease.
This entry is adapted from the peer-reviewed paper 10.3390/pathogens12020335