 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mahmoud Izadi | + 1921 word(s) | 1921 | 2021-11-22 09:11:36 |
Video Upload Options
Research in biomedical sciences has changed dramatically over the past fifty years. There is no doubt that the discovery of apoptosis and autophagy as two highly synchronized and regulated mechanisms in cellular homeostasis are among the most important discoveries in these decades. Along with the advancement in molecular biology, identifying the genetic players in apoptosis and autophagy has shed light on our understanding of their function in physiological and pathological conditions. Apoptosis and autophagy play essential roles in human health, and their malfunction leads to many diseases, including cancer, neurodegenerative disease, and autoimmune disorders. These mechanisms are highly regulated, and there is complex crosstalk between them.
1. Crosstalk between Apoptosis and Autophagy
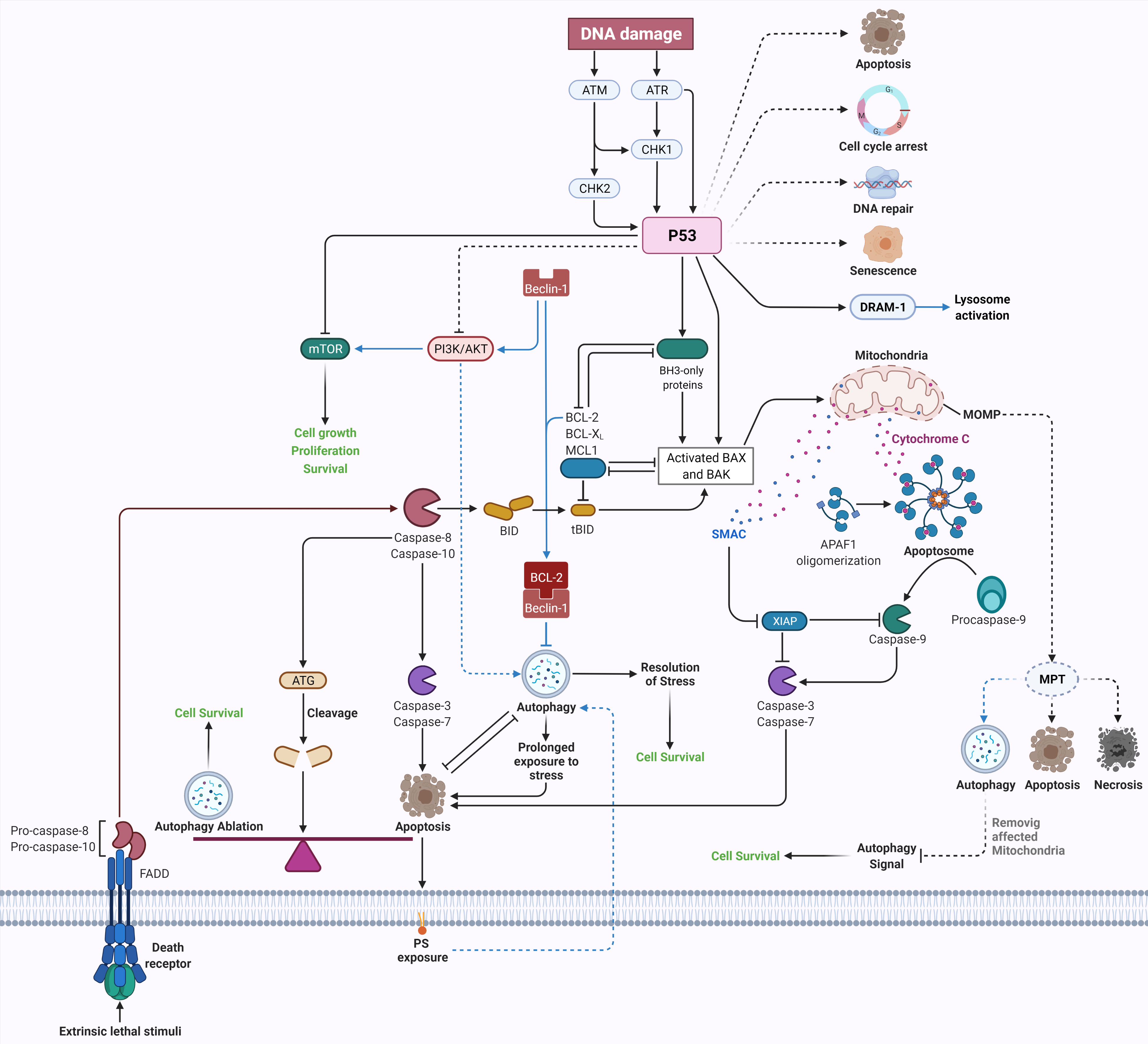
2. Conclusions
References
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687.
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061.
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Marino, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769.
- Yu, X.; Munoz-Alarcon, A.; Ajayi, A.; Webling, K.E.; Steinhof, A.; Langel, U.; Strom, A.L. Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model. J. Mol. NeuroSci. 2013, 50, 586–599.
- Tavernarakis, N.; Pasparaki, A.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. The effects of p53 on whole organism longevity are mediated by autophagy. Autophagy 2008, 4, 870–873.
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134.
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820.
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082.
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742.
- Arienti, S.; Barth, N.D.; Dorward, D.A.; Rossi, A.G.; Dransfield, I. Regulation of Apoptotic Cell Clearance during Resolution of Inflammation. Front. Pharmacol. 2019, 10, 891.
- Yin, C.; Heit, B. Cellular Responses to the Efferocytosis of Apoptotic Cells. Front. Immunol. 2021, 12, 631714.
- Kloditz, K.; Chen, Y.Z.; Xue, D.; Fadeel, B. Programmed cell clearance: From nematodes to humans. BioChem. Biophys. Res. Commun. 2017, 482, 491–497.
- Lemasters, J.J.; Nieminen, A.L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1998, 1366, 177–196.
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. BioChem. Biophys. Res. Commun. 2003, 304, 463–470.
- Vargas, J.L.; Roche, E.; Knecht, E.; Grisolía, S. Differences in the half-lives of some mitochondrial rat liver enzymes may derive partially from hepatocyte heterogeneity. FEBS Lett. 1987, 224, 182–186.
- Pfeifer, U. Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J. Cell Biol. 1978, 78, 152–167.
- Menzies, R.A.; Gold, P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 1971, 246, 2425–2429.
- Dunn, W.A., Jr. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J. Cell Biol. 1990, 110, 1923–1933.
- Elmore, S. Detection of mitochondrial depolarization during autophagy by confocal fluorescence resonance energy transfer (CFRET). Cell Vis. 1997, 4, 170–171.
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301.
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040.
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502.
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal 2014, 26, 549–555.
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975.
- Kapuy, O.; Vinod, P.K.; Mandl, J.; Banhegyi, G. A cellular stress-directed bistable switch controls the crosstalk between autophagy and apoptosis. Mol. Biosyst. 2013, 9, 296–306.
- Lai, M.; Amato, R.; La Rocca, V.; Bilgin, M.; Freer, G.; Spezia, P.; Quaranta, P.; Piomelli, D.; Pistello, M. Acid ceramidase controls apoptosis and increases autophagy in human melanoma cells treated with doxorubicin. Sci. Rep. 2021, 11, 11221.
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056.
- Ojha, R.; Ishaq, M.; Singh, S.K. Caspase-mediated crosstalk between autophagy and apoptosis: Mutual adjustment or matter of dominance. J. Cancer Res. Ther. 2015, 11, 514–524.
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A molecular switch node in the crosstalk between autophagy and apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083.
- Betin, V.M.; Lane, J.D. Atg4D at the interface between autophagy and apoptosis. Autophagy 2009, 5, 1057–1059.
- You, M.; Savaraj, N.; Kuo, M.T.; Wangpaichitr, M.; Varona-Santos, J.; Wu, C.; Nguyen, D.M.; Feun, L. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol. Cell BioChem. 2013, 374, 181–190.
- Djavaheri-Mergny, M.; Maiuri, M.C.; Kroemer, G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 2010, 29, 1717–1719.
- Li, X.; Su, J.; Xia, M.; Li, H.; Xu, Y.; Ma, C.; Ma, L.; Kang, J.; Yu, H.; Zhang, Z.; et al. Caspase-mediated cleavage of Beclin1 inhibits autophagy and promotes apoptosis induced by S1 in human ovarian cancer SKOV3 cells. Apoptosis 2016, 21, 225–238.
- Siddiqui, M.A.; Mukherjee, S.; Manivannan, P.; Malathi, K. RNase L Cleavage Products Promote Switch from Autophagy to Apoptosis by Caspase-Mediated Cleavage of Beclin-1. Int. J. Mol. Sci. 2015, 16, 17611–17636.
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504.
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477.
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell Death Differ. 2017, 24, 1369–1379.
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18.
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566.
- Han, J.; Hou, W.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. A Complex between Atg7 and Caspase-9: A Novel Mechanism of Cross-Regulation Between Autophagy and Apoptosis. J. Biol. Chem. 2014, 289, 6485–6497.
- Sun, Q.; Gao, W.; Loughran, P.; Shapiro, R.; Fan, J.; Billiar, T.R.; Scott, M.J. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J. Biol. Chem. 2013, 288, 15947–15958.
- Lamy, L.; Ngo, V.N.; Emre, N.C.; Shaffer, A.L., 3rd; Yang, Y.; Tian, E.; Nair, V.; Kruhlak, M.J.; Zingone, A.; Landgren, O.; et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 2013, 23, 435–449.
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152.
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371.
- Nah, J.; Zablocki, D.; Sadoshima, J. Autosis: A New Target to Prevent Cell Death. JACC Basic Transl. Sci. 2020, 5, 857–869.
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376.
- Munoz-Pinedo, C.; Martin, S.J. Autosis: A new addition to the cell death Tower of Babel. Cell Death Dis. 2014, 5, e1319.


