Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | fatemeh hajibabaie | -- | 3555 | 2023-12-03 11:22:05 | | | |
| 2 | Rita Xu | Meta information modification | 3555 | 2023-12-04 03:13:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hajibabaie, F.; Abedpoor, N.; Mohamadynejad, P. Types of Cell Death. Encyclopedia. Available online: https://encyclopedia.pub/entry/52285 (accessed on 09 August 2026).
Hajibabaie F, Abedpoor N, Mohamadynejad P. Types of Cell Death. Encyclopedia. Available at: https://encyclopedia.pub/entry/52285. Accessed August 09, 2026.
Hajibabaie, Fatemeh, Navid Abedpoor, Parisa Mohamadynejad. "Types of Cell Death" Encyclopedia, https://encyclopedia.pub/entry/52285 (accessed August 09, 2026).
Hajibabaie, F., Abedpoor, N., & Mohamadynejad, P. (2023, December 03). Types of Cell Death. In Encyclopedia. https://encyclopedia.pub/entry/52285
Hajibabaie, Fatemeh, et al. "Types of Cell Death." Encyclopedia. Web. 03 December, 2023.
Copy Citation
Inflammation and free radicals can stimulate cell self-destruction. Inflammation and cell death are vital aspects of most diseases. Accumulation of cell damage leads to the impairment and dysregulation of the cell function. Thus, understanding the pathomechanism and molecular signaling pathways involved in cell death is necessary.
programmed cell death
uncontrolled cell death
molecular pathways
1. Introduction
Over the course of the past ten years, the nomenclature committee on cell death has dedicated its efforts to the establishment of criteria for the systematic classification and analysis of cell death, encompassing morphological, biochemical, and functional aspects [1][2]. Srinivasan et al. presented a comprehensive analysis of the advancements achieved by computational and systems biologists in elucidating the many regulatory mechanisms involved in cell death. These mechanisms together form the intricate network responsible for controlling cell death processes. The cell death network is characterized as an all-encompassing decision-making process that regulates many biochemical circuits responsible for executing cell death [3]. This network incorporates a variety of feedback and feed-forward loops, as well as the crosstalk across several pathways involved in the regulation of cell death. Indeed, comprehending the intricate dynamics of these intricate regulatory processes necessitates the use of mathematical modeling and system-oriented methodologies [3]. Mathematical modeling functions as a potent instrument for establishing a connection between molecular biology and cell physiology. It achieves this by linking the qualitative and quantitative characteristics of dynamic molecular networks with signal-response curves that are recorded by cell biologists [4]. The dynamics of complex molecular networks that regulate the cell cycle [5][6], nutritional signaling [7], checkpoints [8], signaling dysregulation in cancer [9], and cell death [10][11][12][13] have been effectively described using mathematical and systems-oriented methodologies. Without a doubt, the control of cell death is a molecular process that needs mathematical modeling in order to attain a comprehensive understanding of the process. In a cell’s lifetime, there are four main biological processes: survival, cell division, differentiation, and cell death [2]. Eliminating damaged cells and maintaining the organism’s homeostasis are two of the primary functions of cell death during embryonic development [14]. A cell dies when it stops dividing and functioning as a part of a living organism. This phenomenon may occur because of the body’s normal cellular turnover rate, as a consequence of disorders or localized damage, or as a result of the organism’s death, from which the cells originate [15][16]. There were initially three types of cell death [1] (Figure 1):
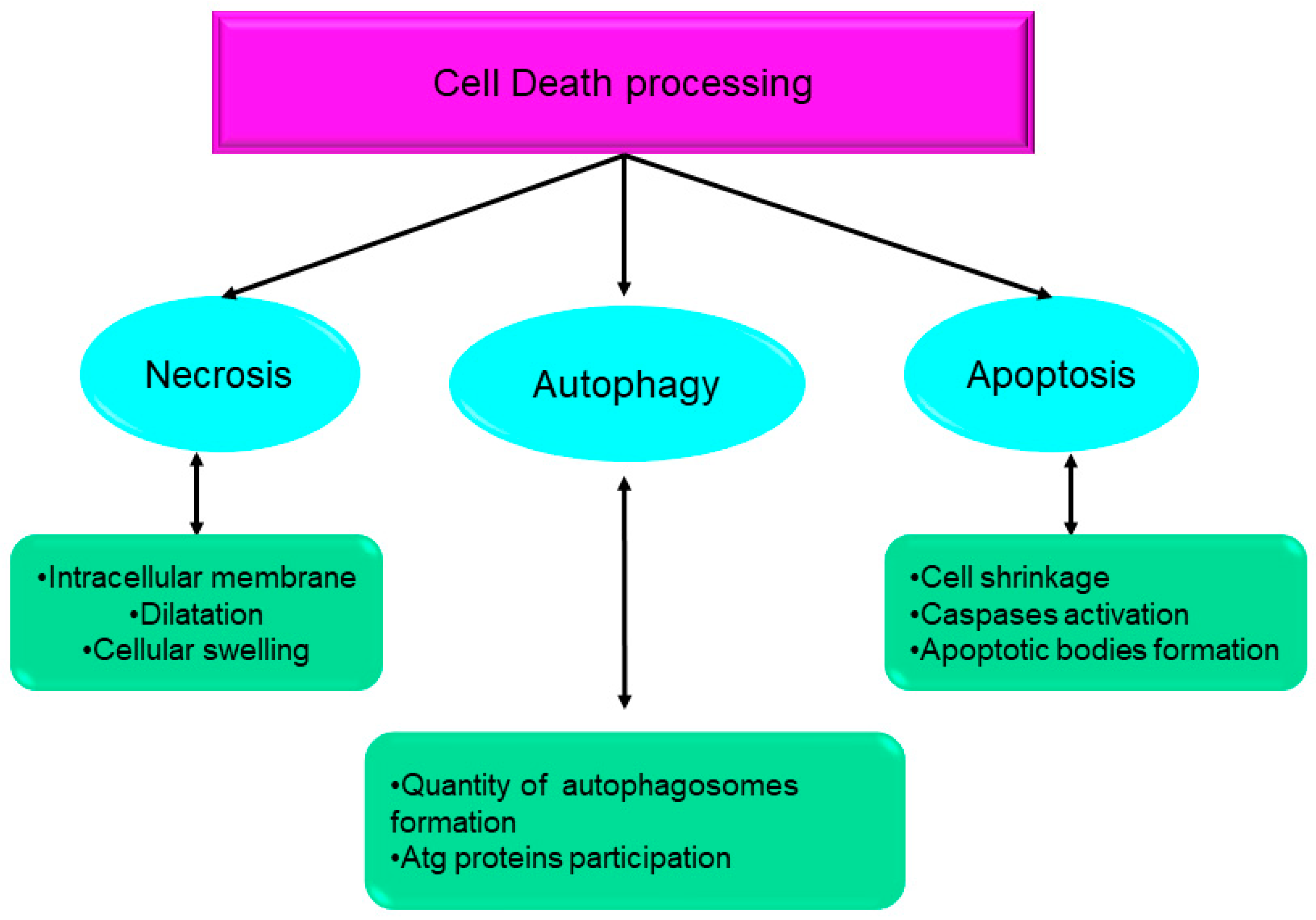
Figure 1. Systematic classification and analysis of cell death, encompassing morphological, biochemical, and functional aspects. Atg: autophagy-related gene.
-
Type I cell death (apoptosis);
-
Type II cell death (autophagy);
-
Type III cell death (necrosis).
Although regulated cell death (RCD) is commonly associated with maintaining organismal homeostasis in both physiological and pathological contexts, it is worth noting that RCD is not exclusive to multicellular organisms. Unicellular eukaryotes, such as Dictyostelium discoideum, and prokaryotic organisms, such as Escherichia coli, also exhibit regulated cell death [17][18]. Moreover, there is instantaneous and catastrophic cell death in contrast to regulated cell death. The cell death category occurs by an exposure to severe physical, chemical, or mechanical attacks [19]. It is worth noting that RCD requires a specialized regulatory network, implying that it can be modulated (accelerated or delayed) by pharmacological intermediation or genetic modification. RCD is implicated in two very different scenarios, despite the underlying molecular pathways showing substantial similarities [20]. RCD may occur without any direct environmental disruption, acting as an established trigger of physiological systems for proliferation or tissue regeneration. RCD may originate from extracellular or intracellular microenvironmental effectors and is disturbed in a way considered as an acute adaptive mechanism for homeostasis offset and stress suppression.
Adaptive stress responses are similar to stress-driven RCD since both aim to maintain a state of biological homeostasis. Despite cellular homeostasis, which controls adaptative stress responses at the cellular level, RCD contributes directly to organism or colony levels. This homeostatic mechanism not only involves the removal of dysfunctional or harmful cells but also allows for the release of chemicals component from dying cells that serve as an early warning system for the other neighboring cells. Common names for these warning signals include damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and alarmins [21][22][23][24].
Cell death strategies may be divided into two groups, programmed and non-programmed cell death, depending on whether they rely on a signal to initiate death [17]. Intracellular signal transduction pathways are the systems that commit cells to programmed cell death (PCD) mechanisms [25]. PCD can be subdivided into non-apoptotic and apoptotic cell death based on morphological properties and molecular interactions [26]. The membrane of the dying cell is conserved during the caspase-dependent process of apoptosis. However, evidence shows that caspases trigger the activation of gasdermin proteins whose N-terminus fragments create pores within the membrane which, upon accumulation, end up causing plasma membrane rupture. For example, caspase-dependent cell death (such as pyroptosis) can cause membrane rupturing as an exception to this classification. In contrast, in scientific terms and based on previous studies, caspase-independent cell death and membrane rupturing are considered non-apoptotic cell death signs [14][25].
When it comes to maintaining a homoeostatic balance in multicellular organisms, the body of an organism continuously attempts to keep the number of new cells formed during mitosis equal to the number of damaged or unnecessarily destroyed cells [27]. Large numbers of regulatory genes are required for controlling cell cycling processes that identify cellular abnormalities and trigger apoptosis, a kind of programmed cell death [14]. Many of these regulatory genes either promote or suppress mitosis, as well as begin apoptosis, autophagy, pyroptosis, and another type of programmed cell death. Diseases like cancer, which may spread throughout an organism and eventually kill it, result from uncontrolled cell division [28]. In contrast, degenerative statuses such as rheumatoid arthritis, Parkinson’s, and Alzheimer’s result from excessive cell death rates [29][30]. In light of the extensive and complex interplay of RNAs and proteins inside the cellular processes of the cell cycle and cell death, some regulatory proteins, receptors, and enzymes have been identified as key regulators. Mutations or aberrant expression of these regulators may directly affect the cell cycle machinery [31][32].
Distinct macroscopic morphological changes accompany the death of cells. The utilization of morphotypes has been employed to categorize cell death into three distinct categories, predicated with the methodologies employed for the elimination of diseased cells and their fragments [33].
2. Types of Cell Death
2.1. Apoptosis
Apoptosis or Type I cell death is associated with the following cellular events:
-
Cytoplasmic shrinkage;
-
The irreversible condensation of chromatin in the nucleus (pyknosis);
-
The destructive fragmentation of the nucleus (karyorrhexis);
-
The formation of apoptotic bodies based on the establishment of intact small vesicles;
-
The phagocytosis and decomposition of apoptotic bodies in neighboring cells’ lysosomes [34].
In a publication in 1972, Kerr, Wyllie, and Currie proposed the word “Apoptosis” to characterize a specific form of cell death [35]. Initiating the apoptosis process is associated with the stopping of the proliferation and division of the cell. In contrast, the cellular entity undergoes a regulated mechanism resulting in its demise, while the intracellular contents remain contained within the confines of the cellular milieu. Apoptosis is recognized as a cellular suicide process that is established by triggering a series of cysteine-aspartic proteases that is term as the caspases activation cascade. Caspases may be divided into two classes: those that act as “Initiators Caspase” and those that act as “Executioners Caspase” [36] (Figure 2).
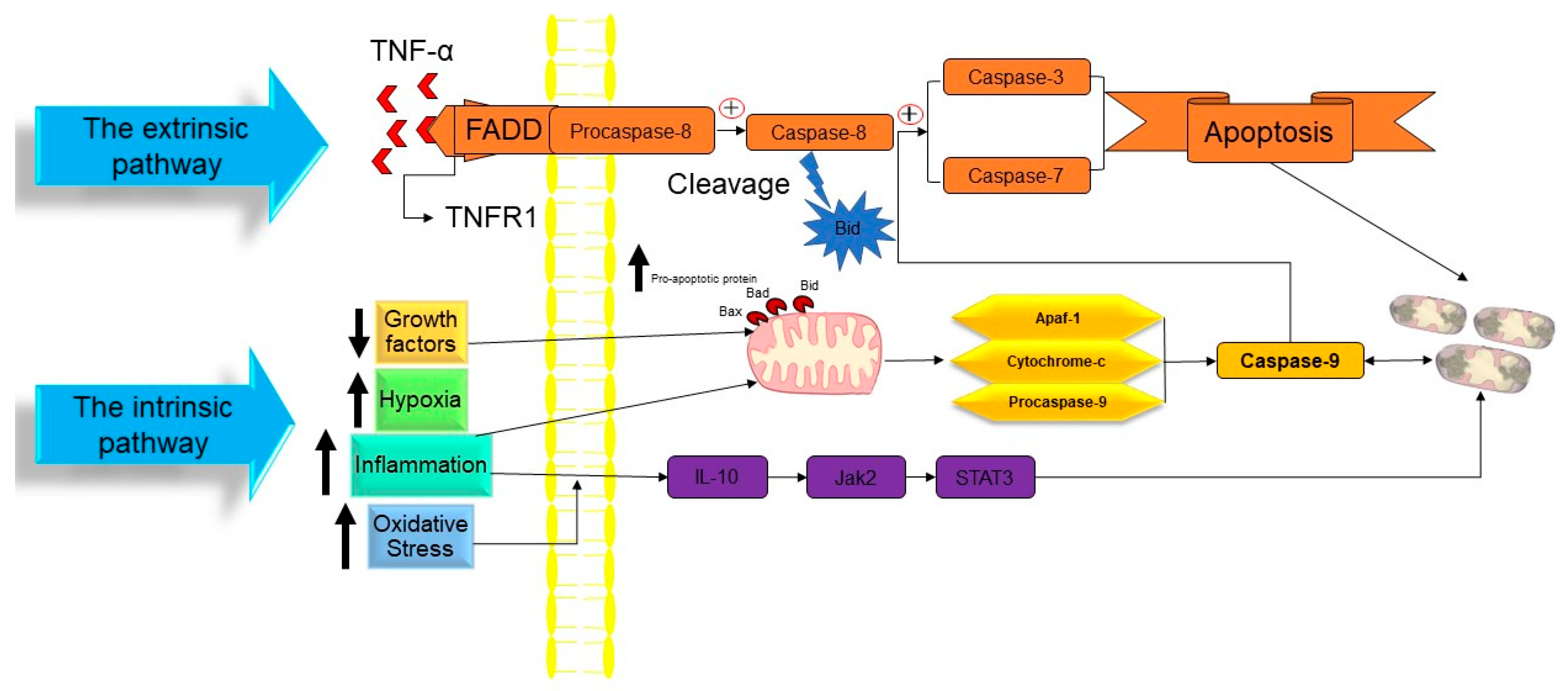
Figure 2. The molecular mechanism of apoptotic cell death. TNF: tumor necrosis factor, TNFR1: tumor necrosis factor receptor 1, FADD: FAS-associated death domain, Bid: BH3 interacting domain death agonist, Bad: BCL2-associated agonist of cell death, Bax: Bcl-2-associated X protein, Apaf1: apoptotic protease activating factor-1, IL-10: interleukin 10, Jak2: janus kinase 2, and STAT3: signal transducer and activator of transcription 3. ↑ Indicated increasing of the factor. ↓ Indicated reducing of the factor.
At the time cell damage is sensed, initiator procaspases 8 and 9 are converted into active initiator caspases, and consequently, they induce the executioner caspases’ activation (caspases 3, 6, and 7). An array of processes occur in apoptotic bodies’ production and destruction of damaged cells, including DNA fragmentation, telomeres’ shortening, the degradation of proteins/cytoskeleton/crosslinking of a protein, phagocytic cell ligands’ expression, and apoptotic body formation [37]. As a genetically conserved mechanism, apoptosis is highly regulated in multicellular organisms. There are two pathways for the apoptosis process:
-
The intrinsic pathway based on intracellular damage sensors’ detection.
-
The extrinsic pathway based on immune cell and damaged cell attachment.
The apoptotic cell death rate in humans is around 1 × 109 cells/day [38].
-
The intrinsic pathway of apoptosis
The intrinsic route of apoptosis, also referred to as the mitochondrial pathway, involves the interaction of many stimuli with diverse cellular targets. [39]. This kind of apoptosis is triggered by either a positive and/or negative pathway and relies on substances produced by the mitochondria [40]. A deficiency of growth factors, cytokines, and hormones in the cellular microenvironment might trigger a negative signal to initiate the apoptosis process [14]. Apoptosis is triggered by activating pro-apoptotic molecules like p53 upregulated modulator of apoptosis (Puma), Noxa, and Bcl-2-associated X (Bax) without pro-survival signals [41]. Apoptosis may also be triggered by an exposure to positive variables such as hypoxia, poisons, radiation, reactive oxygen species (ROS), viruses, and other hazardous agents, yet in the case of certain cells, like neutrophils, hypoxia can enhance cell survival [42]. Mitochondrial outer membrane permeabilization (MOMP) is a pivotal stage in this process. The modulation of MOMP is influenced by the activity of several pro-apoptotic and anti-apoptotic members belonging to the BCL2 protein family, which serves as an apoptosis regulator. Upon exposure to an apoptotic stimuli, the mitochondrial outer membrane permeabilization process is triggered, resulting in the sequential activation of the initiator caspase 9 (CASP9) followed by the executioner caspases CASP3 and CASP7 [43]. Researchers have successfully discovered two separate classes of pro-apoptotic BCL2 proteins that exhibit different functional characteristics. The first category comprises the apoptotic activators BAX, BAK1, and BOK. Upon being triggered by apoptotic signals, BAX, BAK1, and BOK initiate MOMP by creating holes in the outer mitochondrial membrane (OMM). These pro-apoptotic factors stimulate the release of many apoptogenic substances such as Cytochrome-C and diablo IAP-binding mitochondrial protein (DIABLO; also known as second mitochondrial activator of caspases, SMAC) into the cytoplasm. The apoptogenic activity of Cytochrome-C is manifested by its interaction with apoptotic peptidase activating factor 1 (APAF1) and pro-CASP9, resulting in the formation of a complex referred to as the apoptosome. This complex then triggers the sequential activation of CASP9, as well as of the executioner caspases CASP3 and CASP7. The activation of CASP3 and CASP7 is facilitated by the interaction of DIABLO/SMAC with X-linked inhibitor of apoptosis (XIAP) and other members that are inhibitors of the apoptosis (IAP) protein family [43]. The second category of pro-apoptotic BCL2 proteins, referred to as BH3-only proteins, encompasses a group of molecules including BAD, PUMA, BIK, BIM, BMF, BID, HRK, and NOXA. Direct interaction between caspase-cleaved BID (tBID), BIM, PUMA, and NOXA in the mitochondria has the capability to facilitate the activation of BAX and BAK1. On the other hand, indirect activation of BAX and BAK1 occurs when BH3-only proteins including BAD, BIK, BMF, and HRK bind to and inhibit the activity of anti-apoptotic BCL2 family members [44][45]. Caspase-9 is the initiator caspase that regulates the intrinsic mechanism of apoptosis by binding to the apoptotic protease activating factor-1 (APAF1) once its caspase recruitment domains (CARD) have been exposed [14][46]. In nonactive apoptotic cells, APAF1 is often folded to prevent its CARD domain from binding to procaspase-9 [47]. The interaction between Cytochrome-C and the tryptophan-aspartic acid (WD) domain of APAF1 monomers induces a structural change in APAF1, leading to the exposure of a region responsible for nucleotide binding and oligomerization. This region specifically binds to deoxyadenosine triphosphate (dATP), hence triggering the initiation of apoptosis [48]. Due to the extra conformational shift induced by this interaction, the CARD and oligomerization domains of APAF1 are exposed, allowing for the assembly of several APAF1s into an apoptosome [49]. Many procaspase-9 proteins are recruited and activated by the apoptosome’s exposed CARD domains, which are located in the open core of the cell death complex [14]. When the caspase 9 is activated, it triggers the executioner procasp-3, which, once converted to active caspase-3, causes the complete induction of apoptosis. However, gasdermin E, a substrate of active caspase-3, induces pyroptosis, rather than apoptosis. While apoptosis may be triggered by the actions of Smac/Diablo and HtrA2/Omi, the inhibition of inhibitors of apoptosis proteins (IAPs) is inadequate without the release of Cytochrome-C [50][51].
-
Extrinsic pathway of apoptosis
Extrinsic apoptosis, also called the death receptor (DR) pathway, is triggered when death ligands are released by patrolling NK-cells or macrophages and bind with DRs on the target cell membrane [52]. This triggers the extrinsic route, which in turn activates caspase 8 from pro-caspase-8. The DRs are proteins with structural and functional similarities with the tumor necrosis factor (TNF) superfamily [53][54]. When a death ligand binds to a DR, the DR’s cytoplasmic domain becomes a death-inducting signaling complex (DISC), where monomeric pro-caspase-8 is recruited through its death effector domain (DED) [55][56]. The adaptor protein known as the TNFR-associated death domain (TRADD), or the FAS-associated death domain (FADD), is also part of the DISC. It aids in the binding of pro-caspase-8 [57]. Multiple pro-caspase-8 monomers are recruited to the DISC, where they undergo dimerization and trigger to become caspase-8. Caspase-8 may initiate an apoptosis mechanism by any of two sub-pathways [14][58].
Whether the cells are type I or type II determines which sub-pathway gets activated [59]. Caspase-8 directly cleaves executioner caspases, triggering apoptosis in type I cells [60]. Unless blocked by proteins secreted from the mitochondria, IAPs prevent the direct activation of the executioner caspases by caspase-8. Mice lacking caspase-8, who normally respond to DR ligands, reveal the crucial function that caspase-8 plays in the regulation of the apoptosis extrinsic cascade [51]. Whether apoptosis is initiated by the intrinsic or extrinsic routes, it must be tightly regulated to avoid the disastrous outcomes that might result from insufficient control. Mutations in the multiple apoptosis initiation systems are a common cause of cancer [61][62]. The creation of a benign tumor or cancer results from this phenomenon when it happens in conjunction with a failure to react to external cues that would ordinarily trigger the extrinsic route or prevent proliferation [63].
2.2. Anoikis
For the first time, Frisch described the “Anoikis” concept in 1994 [64]. The loss of integrin-dependent anchoring is considered to be the trigger for the anoikis subtype of intrinsic apoptosis [65]. Anoikis, a Greek word meaning “homelessness” or “loss of home,” describes the one type of apoptosis that occurs when cells lose their connection to the extracellular matrix (ECM) and adhere to an unsuitable site [66]. Integrin receptors are mediators of ECM interaction and are essential for migration, proliferation, and survival because they not only establish physical linkages with the cytoskeleton but also transduce signals from the ECM to the cell [67]. By preventing cells from detaching and re-adhering to inappropriate matrices, as well as by inhibiting dysplastic development, anoikis serves as a vital defensive mechanism for an organism [68][69]. Because of this, adherent cells may be able to survive in a suspension or proliferate in ectopic places where the ECM proteins are different if the anoikis program is not well executed [70]. Emerging evidence suggests that cancer cells’ aberrant execution of anoikis is a characteristic of the disease that promotes metastasis to distant organs [65]. Anoikis is a kind of apoptosis that is produced by insufficient or incorrect ECM connections but otherwise follows the same mechanisms as apoptosis [69][71].
The induction and execution of anoikis include many routes that converge on the activation of caspases and subsequent molecular processes. This activation triggers the activation of endonucleases, resulting in DNA fragmentation and ultimately leading to cell death. [72]. Two apoptotic routes, the intrinsic pathway involving mitochondrial dysfunction and the extrinsic pathway involving the activation of cell surface death receptors, work together to initiate the anoikis program (the extrinsic pathway) [65]. Proteins belonging to the B cell lymphoma-2 (Bcl-2) family play important roles in both of these processes. Three subfamilies exist within the Bcl-2 family, and they are as follows [66]:
- A.
-
Myeloid cell leukemia sequence 1, as well as the anti-apoptotic proteins Bcl-2 and B-cell lymphoma-extra large, Bcl-XL (Mcl-1).
- B.
-
Pro-apoptotic proteins Bax, Bcl-2 homologous antagonist/killer (Bak), and Bcl-2 related ovarian killer (Bok), all with several domains.
- C.
-
BH3 interacting domain death agonist (Bid), BCL2-associated agonist of cell death (Bad), Bcl-2 interacting mediator of cell death (Bim), BCL-2 interacting killer (Bik), BCL-2 modifying factor (Bmf), Noxa, Puma, and Harakiri (Hrk) are all pro-apoptotic BH3-only proteins [66].
-
The intrinsic pathway of Anoikis
DNA damage and endoplasmic reticulum stress are two intracellular cues that initiate the intrinsic pathway of apoptosis. In this process, mitochondria play a crucial role in regulating apoptosis [73]. In response to death signals, the pro-apoptotic proteins Bax and Bak undergo translocation from the cytosol to the outer mitochondrial membrane (OMM). The oligomerization of these proteins leads to the formation of a channel through the OMM, which in turn causes mitochondrial permeabilization. This permeabilization event subsequently triggers the release of Cytochrome-C [74]. In addition to the Bax proteins’ inherent pore-generating activity, their interaction with mitochondrial channel proteins, such as the voltage-dependent anion channels, may also contribute to membrane permeabilization [65]. The activation of the effector caspase-3 occurs subsequent to the release of Cytochrome-C. Apoptosis is initiated by assembling the apoptosome complex, which then triggers the activation of caspase-9 and the cofactor APAF [75]. The BH3-only pro-apoptotic proteins are known to have significant functions in the intrinsic pathway of the anoikis cell death mechanism. Bid and Bim are proteins from this biological family that become active upon cellular detachment from the ECM, hence enhancing the production of Bax-Bak oligomers within the OMM [76]. “Activators” refers to this class of BH3-only proteins [77]. Specifically, Bim is confined to the dynein cytoskeletal complexes until cell separation triggers its release and translocation to the mitochondria [78][79].
The process of Bim phosphorylation by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) is initiated during integrin contact. This phosphorylation event serves to impede the proteasomal degradation of Bim, resulting in its accumulation subsequent to the loss of cell adhesion [80]. Bik, Bmf, Bad, Puma, Hrk, and Noxa are all examples of so-called “Sensitizers”, another class of BH3-only proteins [65]. The inhibitory impact of Bcl-2 on apoptosis is impeded when sensitizer BH3-only proteins engage in a competition for its BH3 binding domain. This competition enables activator BH3-only proteins to initiate the development of Bax-Bak oligomers, hence facilitating cellular demise [81]. The Bmf functions as a surveillance mechanism in epithelial cells, detecting disruptions in the cytoskeleton structure and transmitting signals that initiate cell death [82]. Following the process of cell separation, Bmf becomes dissociated from the myosin V motor complex and subsequently accumulates within the mitochondria. Within this organelle, Bmf acts to counteract the effects of Bcl-2, thus initiating the release of Cytochrome-C and ultimately leading to the execution of anoikis [65].
-
The extrinsic pathway of Anoikis
The execution of anoikis involves the activation of both the intrinsic and extrinsic pathways. The initiation of the DISC occurs in the extrinsic route when a ligand binds to death receptors belonging to the TNFR superfamily. These death receptors include the Fas receptor, TNF receptor superfamily 1 (TNFR1) receptor, and the TNF-related apoptosis-inducing ligand (TRAIL) receptors-1 and TRAIL receptors-2. DISC, by means of engaging with adaptor proteins such as FADD, facilitates the aggregation of numerous caspase-8 molecules and triggers their subsequent activation [83][84]. Substrate proteolysis and cell death result from the active caspase-8 being secreted into the cytoplasm, where it cleaves and activates the effector caspases, caspases-3, caspases-6, and caspases-7 [77][85]. An alternative mechanism that connects the extrinsic and intrinsic pathways is the cleavage and activation of Bid upon Caspase-8 activation [86][87]. This t-Bid version can induce mitochondrial Cytochrome-C release and apoptosome assembly. Detachment of cells from the ECM has been shown to trigger the release of a mitochondrial protein called Bit1 into the cytoplasm, where it functions as a pro-apoptotic mediator and induces a caspase-independent type of apoptosis [65]. Mitochondrial damage in certain cases is a subsequent effect of the occurrence of death receptor activation, which may be created as a feedback loop between extrinsic death signals and the intrinsic route. Prior studies have demonstrated the significance of the extrinsic pathway in the occurrence of anoikis, wherein the detachment from the ECM triggers the increased expression of Fas and Fas ligand, while simultaneously reducing the levels of FADD-like interleukin-1β-converting enzyme-like inhibitory protein (FLIP), an inherent inhibitor of Fas-mediated signaling [88]. Morphological alterations in cells are another interesting trigger for the extrinsic apoptosis pathway. The rounded shape of a cell, after its detachment has occurred, may cause “induced proximity” of Fas receptors, triggering their activation [65]. Cell death occurs through the convergence of both the extrinsic and intrinsic apoptotic pathways, which are dependent on the activation of the effector caspase-3. The activation of caspase-3 triggers a subsequent proteolytic cascade and exerts an influence on the cellular apoptosis pathway. The cleavage of signaling molecules, such as focal adhesion kinase (FAK) and protein 130 kDa Crk-associated substrate (p130Cas), is of utmost importance for the effective implementation of apoptosis [89][90]. When FAK is cleaved by caspases, it interferes with the structure of focal adhesions and dampens the survival signal they provide. A caspase-mediated cleavage disrupts the localization and interactions of p130Cas with paxillin as an SH2/SH3 adaptor protein that binds to FAK and transmits integrin signals [91]. On the other hand, the C-terminal inhibitory fragment generated by p130Cas cleavage hinders the transcription of p21Waf1/Cip1. Hence, this inhibitory fragment triggers an apoptotic response and also a cell cycle arrest [65].
References
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11.
- Yan, G.E.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features. World Acad. Sci. J. 2020, 2, 39–48.
- Srinivasan, M.; Clarke, R.; Kraikivski, P. Mathematical Models of Death Signaling Networks. Entropy 2022, 24, 1402.
- Tyson, J.J.; Novak, B. A dynamical paradigm for molecular cell biology. Trends Cell Biol. 2020, 30, 504–515.
- Kraikivski, P.; Chen, K.C.; Laomettachit, T.; Murali, T.M.; Tyson, J.J. From START to FINISH: Computational analysis of cell cycle control in budding yeast. NPJ Syst. Biol. Appl. 2015, 1, 15016.
- Shafiekhani, S.; Kraikivski, P.; Gheibi, N.; Ahmadian, M.; Jafari, A.H. Dynamical analysis of the fission yeast cell cycle via Markov chain. Curr. Genet. 2021, 67, 785–797.
- Jalihal, A.P.; Kraikivski, P.; Murali, T.M.; Tyson, J.J. Modeling and analysis of the macronutrient signaling network in budding yeast. Mol. Biol. Cell 2021, 32, ar20.
- Jung, Y.; Kraikivski, P.; Shafiekhani, S.; Terhune, S.S.; Dash, R.K. Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: Computational modeling and analysis. NPJ Syst. Biol. Appl. 2021, 7, 46.
- Clarke, R.; Kraikivski, P.; Jones, B.C.; Sevigny, C.M.; Sengupta, S.; Wang, Y. A systems biology approach to discovering pathway signaling dysregulation in metastasis. Cancer Metastasis Rev. 2020, 39, 903–918.
- Xu, F.; Yin, Z.; Zhu, L.; Jin, J.; He, Q.; Li, X.; Shuai, J. Oscillations governed by the incoherent dynamics in necroptotic signaling. Front. Phys. 2021, 9, 726638.
- Zhu, L.; Li, X.; Xu, F.; Yin, Z.; Jin, J.; Liu, Z.; Qi, H.; Shuai, J. Network modeling-based identification of the switching targets between pyroptosis and secondary pyroptosis. Chaos Solitons Fractals 2022, 155, 111724.
- Konstorum, A.; Tesfay, L.; Paul, B.T.; Torti, F.M.; Laubenbacher, R.C.; Torti, S.V. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 2020, 493, 110222.
- Checcoli, A.; Pol, J.G.; Naldi, A.; Noel, V.; Barillot, E.; Kroemer, G.; Thieffry, D.; Calzone, L.; Stoll, G. Dynamical Boolean Modeling of Immunogenic Cell Death. Front. Physiol. 2020, 11, 590479.
- D’arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366.
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410.
- Galluzzi, L.; López-Soto, A.; Kumar, S.; Kroemer, G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 2016, 44, 221–231.
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164.
- Galluzzi, L.; Pedro, B.-S.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73.
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375.
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875.
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788.
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.M.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Kutscher, L.M.; Shaham, S. Non-apoptotic cell death in animal development. Cell Death Differ. 2017, 24, 1326–1336.
- Cooper, J.P.; Youle, R.J. Balancing cell growth and death. Curr. Opin. Cell Biol. 2012, 24, 802.
- Liao, M.; Qin, R.; Huang, W.; Zhu, H.-P.; Peng, F.; Han, B.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: A revisited perspective from molecular mechanisms to targeted therapies. J. Hematol. Oncol. 2022, 15, 44.
- Jellinger, K.A. Cell death mechanisms in neurodegeneration. J. Cell. Mol. Med. 2001, 5, 1–17.
- Agid, Y.; Blin, J. Nerve cell death in degenerative diseases of the central nervous system: Clinical aspects. Ciba Found. Symp. 1987, 126, 3–29.
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636.
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88.
- Van Cruchten, S.; Van Den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 2002, 31, 214–223.
- Yadav, A.B.; Angadi, P.V.; Kale, A.D.; Yadav, S.K. Histological assessment of cellular changes in postmortem gingival specimens for estimation of time since death. J. Forensic Odonto-Stomatol. 2015, 33, 19.
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180.
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070.
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22.
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180.
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-only proteins noxa and puma are key regulators of induced apoptosis. Life 2022, 12, 256.
- Lee, J.; Song, C.-H. Effect of reactive oxygen species on the endoplasmic reticulum and mitochondria during intracellular pathogen infection of mammalian cells. Antioxidants 2021, 10, 872.
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L. Apoptotic Cell Death in Disease—Current Understanding of the Nccd 2023. Cell Death Differ. 2023, 1–58.
- Kale, J.; Osterlund, E.J.; Andrews, D.W. Bcl-2 Family Proteins: Changing Partners in the Dance Towards Death. Cell Death Differ. 2018, 25, 65–80.
- Chen, H.-C.; Kanai, M.; Inoue-Yamauchi, A.; Tu, H.-C.; Huang, Y.; Ren, D.; Kim, H.; Takeda, S.; Reyna, D.E.; Chan, P.M. An Interconnected Hierarchical Model of Cell Death Regulation by the Bcl-2 Family. Nat. Cell Biol. 2015, 17, 1270–1281.
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65.
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214.
- Green, D.R. The mitochondrial pathway of apoptosis Part I: MOMP and beyond. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038.
- Ferraro, E.; Fuoco, C.; Strappazzon, F.; Cecconi, F. Apoptosome structure and regulation. In Apoptosome: Up-and-Coming Therapeutical Tool; Springer: Berlin/Heidelberg, Germany, 2010; pp. 27–39.
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432.
- Ekert, P.G.; Vaux, D.L. The mitochondrial death squad: Hardened killers or innocent bystanders? Curr. Opin. Cell Biol. 2005, 17, 626–630.
- Shliapina, V.L.; Yurtaeva, S.V.; Rubtsova, M.P.; Dontsova, O.A. At the crossroads: Mechanisms of apoptosis and autophagy in cell life and death. Acta Naturae 2021, 13, 106.
- Schultz, D.R.; Harringto, W.J., Jr. Apoptosis: Programmed cell death at a molecular level. In Seminars in Arthritis and Rheumatism; WB Saunders: Philadelphia, PA, USA, 2003; pp. 345–369.
- Raducka-Jaszul, O.; Bogusławska, D.M.; Jędruchniewicz, N.; Sikorski, A.F. Role of extrinsic apoptotic signaling pathway during definitive erythropoiesis in normal patients and in patients with β-thalassemia. Int. J. Mol. Sci. 2020, 21, 3325.
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural determinants of DISC function: New insights into death receptor-mediated apoptosis signalling. Pharmacol. Ther. 2013, 140, 186–199.
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656.
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355.
- Tan, C.-P.; Lu, Y.-Y.; Ji, L.-N.; Mao, Z.-W. Metallomics insights into the programmed cell death induced by metal-based anticancer compounds. Metallomics 2014, 6, 978–995.
- Samraj, A.K.; Keil, E.; Ueffing, N.; Schulze-Osthoff, K.; Schmitz, I. Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J. Biol. Chem. 2006, 281, 29652–29659.
- Engels, I.H.; Stepczynska, A.; Stroh, C.; Lauber, K.; Berg, C.; Schwenzer, R.; Wajant, H.; Jänicke, R.U.; Porter, A.G.; Belka, C. Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene 2000, 19, 4563–4573.
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 147–171.
- Fadeel, B.; Orrenius, S. Apoptosis: A basic biological phenomenon with wide-ranging implications in human disease. J. Intern. Med. 2005, 258, 479–517.
- Long, J.S.; Ryan, K.M. New frontiers in promoting tumour cell death: Targeting apoptosis, necroptosis and autophagy. Oncogene 2012, 31, 5045–5060.
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626.
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2013, 1833, 3481–3498.
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439.
- Giancotti, F.G. Complexity and specificity of integrin signalling. Nat. Cell Biol. 2000, 2, E13–E14.
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364.
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562.
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393.
- Li, S.; Chen, Y.; Zhang, Y.; Jiang, X.; Jiang, Y.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Shear stress promotes anoikis resistance of cancer cells via caveolin-1-dependent extrinsic and intrinsic apoptotic pathways. J. Cell. Physiol. 2019, 234, 3730–3743.
- Grossmann, J. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis 2002, 7, 247–260.
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163.
- Bras, M.; Queenan, B.; Susin, S.A. Programmed cell death via mitochondria: Different modes of dying. Biochemistry 2005, 70, 231–239.
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080.
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740.
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241.
- Puthalakath, H.; Villunger, A.; O’Reilly, L.A.; Beaumont, J.G.; Coultas, L.; Cheney, R.E.; Huang, D.C.S.; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293, 1829–1832.
- Zhi, Z.; Ouyang, Z.; Ren, Y.; Cheng, Y.; Liu, P.; Wen, Y.; Shao, Y. Non-canonical phosphorylation of Bmf by p38 MAPK promotes its apoptotic activity in anoikis. Cell Death Differ. 2022, 29, 323–336.
- Qi, X.-J.; Wildey, G.M.; Howe, P.H. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem. 2006, 281, 813–823.
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535.
- McConkey, D.J.; Bondar, V. Regulation and function of detachment-induced cell death (Anoikis) in cancer progression and metastasis. Apoptosis Senescence Cancer 2007, 109–122.
- Bunek, J.; Kamarajan, P.; Kapila, Y.L. Anoikis mediators in oral squamous cell carcinoma. Oral Dis. 2011, 17, 355–361.
- Rosen, K.; Shi, W.; Calabretta, B.; Filmus, J. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of fas ligand: A novel mechanism of anoikis of intestinal epithelial cells. J. Biol. Chem. 2002, 277, 46123–46130.
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636.
- Mühlethaler-Mottet, A.; Bourloud, K.B.; Auderset, K.; Joseph, J.-M.; Gross, N. Drug-mediated sensitization to TRAIL-induced apoptosis in caspase-8-complemented neuroblastoma cells proceeds via activation of intrinsic and extrinsic pathways and caspase-dependent cleavage of XIAP, Bcl-xL and RIP. Oncogene 2004, 23, 5415–5425.
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2011, 1813, 558–563.
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-flip and implications for anoikis. J. Cell Biol. 2001, 152, 633–644.
- Alahari, S.K.; Reddig, P.J.; Juliano, R.L. Biological aspects of signal transduction by cell adhesion receptors. Int. Rev. Cytol. 2002, 220, 145–184.
- Shim, S.R.; Kook, S.; Kim, J.I.; Song, W.K. Degradation of focal adhesion proteins paxillin and p130cas by caspases or calpains in apoptotic rat-1 and L929 cells. Biochem. Biophys. Res. Commun. 2001, 286, 601–608.
- Kim, W.; Kook, S.; Kim, D.J.; Teodorof, C.; Song, W.K. The 31-kDa caspase-generated cleavage product of p130cas functions as a transcriptional repressor of E2A in apoptotic cells. J. Biol. Chem. 2004, 279, 8333–8342.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Revisions:
2 times
(View History)
Update Date:
04 Dec 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No