 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tomas Horsten | + 3708 word(s) | 3708 | 2021-06-08 08:39:15 | | | |
| 2 | Karina Chen | Meta information modification | 3708 | 2021-06-09 10:00:57 | | |
Video Upload Options
Dihydropyrrolo[1,2-a]pyrazinone rings are a class of heterocycles present in a wide range of bioactive natural products and analogues thereof. As a direct result of their bioactivity, the synthesis of this privileged class of compounds has been extensively studied. This review provides an overview of these synthetic pathways.
1. Introduction
Nitrogen-containing heteroaromatic rings are valuable motifs in bioactive molecules and recurrent scaffolds present in drugs [1][2]. The application of nitrogen ring systems in drug development is related to their diverse properties, including relatively small conformational freedom, while retaining some polarity, compared to aromatic hydrocarbons. Additionally, commercial availability, synthetic tractability, chemical diversity and the tendency for functionalization should also be highlighted [3]. However, the wide chemical space of nitrogen heterocycles is not yet fully explored in the attempt to find new drug candidates.
Dihydropyrrolo[1,2-a]pyrazinone rings are found in the structure of a number of bioactive compounds, including synthetic and natural products isolated from various sources like fungi, plants or sponges. These natural products (some structures are shown in Figure 1) often contain one or two bromine substituents on the pyrrole ring. The simplest congeners are longamide A [4] and its nonbrominated analog mukanadin C [5] (not shown), longamide B [6], hanishin [7], stylisine D [8], cyclooroidin [9], and agesamide [10]. More complicated tetracyclic analogs include dibromophakellstatin, dibromophakellin [11] and the different agelastatins A-F [12][13]. One of the most complicated pyrrolopyrazinone natural products is palau’amine [14], and its structure has been seen as a challenge for total synthesis. Some related natural products are the higher oxidation state analogs peramine [15] and nannozinone B [16], containing the pseudoaromatic pyrazinone ring, the pyrrolodiketopiperazines brevianamide T [17] and macrophominol [18], and the oxopyrrole derivative oxocyclostylidol [19] (Figure 1).
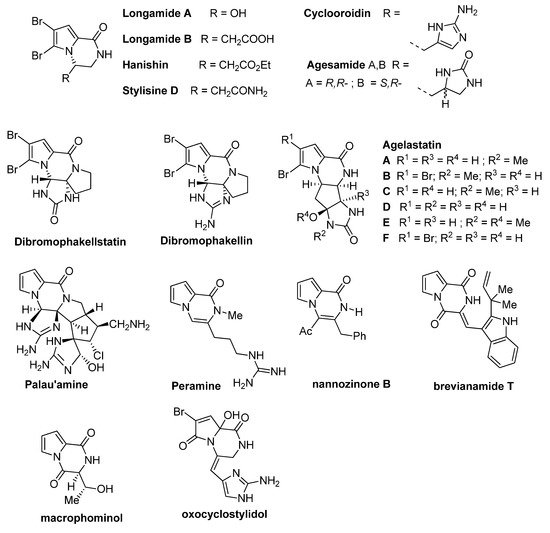
Figure 1. Pyrrolopyrazinone natural products.
Several bioactivities have been found for these pyrrolopyrazinone natural products. Hanishin shows cytotoxicity against non-small cell lung carcinoma [7], and agelastatin A and D display significant activity against different cell lines [20]. Longamide B was found to have antiprotozoal [21] and antibacterial [6] properties, with good potency against African trypanosome. Palau’amine and the similar dibromophakellin and dibromophakellstatin inhibit the human 20S proteasome [22]. Peramine is an insect feeding deterrent [23].
2. Fusion of a Pyrazinone to a Pyrrole Derivative
2.1. Starting from 2-Monosubstituted Pyrroles
The most common way toward pyrrolopyrazinones is fusing a pyrazinone to a pyrrole. One way to realize this is starting from 1H-pyrrole-2-carboxamide bearing electrophilic groups on the amide that react in an intramolecular fashion with the nucleophilic pyrrole nitrogen. Several electrophilic groups are possible. Electron-poor alkenes can undergo aza-Michael addition, as in the base-catalyzed formation of N-benzyl longamide B derivative 8 from the corresponding open chain pyrrole-2-amide 7 after potassium carbonate (K2CO3)-catalyzed cyclization, bromination with N-bromosuccinimide (NBS) and saponification (Scheme 1) [24]. A similar aza-Michael cyclization was reported in the total synthesis of longamide B and cyclooroidin via the Wadsworth–Horner–Emmons olefination of longamide A [25][26] or in the 1,8-diazabicyclo[5.4.0]undec-7-ene(DBU)-catalyzed cyclization of precursors to kinase inhibitors 5 [27]. An enantioselective aza-Michael cyclization (up to 56%ee) was realized with compounds analogous to 7 in the presence of a chiral N-benzylammonium phase transfer catalyst derived from quinine [28].
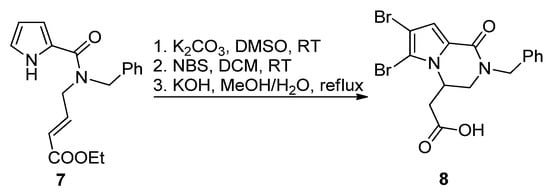
Scheme 1. Aza-Michael reaction leading to a longamide B derivative.
The total synthesis of (-)-agelastatin A involved a similar aza-Michael reaction to an enone intermediate 9, which was generated by the oxidation of an allylic alcohol precursor [29] or by a metathesis reaction [30]. Different bases were tried for the cyclization of 9 to the intermediate 10 that then could be further elaborated to the natural product. It was found that diisopropylethylamine (DIPEA) in THF is a suitable base/solvent combination after the acidity of the pyrrole is increased by bromination, whereas nonbrominated pyrrole 9 resulted in the recovery of the starting material, rearrangement and/or decomposition [29][31][32]. Many variants of this cyclization have been described, with other base/solvent combinations like cesium carbonate in methanol [30] or THF [33] at room temperature, potassium carbonate in dimethyl sulfoxide (DMSO) at 100 °C [34], trimethylamine in acetonitrile (ACN) at −20 °C [35] and triethylamine (Et3N) in DMSO at room temperature with the in situ generation of enone 9 by the elimination of a sulfone group [36][37] (Scheme 2).
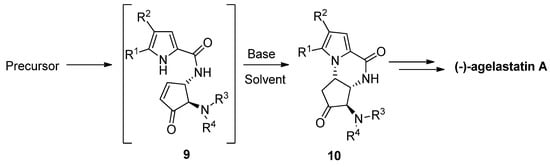
Scheme 2. Aza Michael reaction as part of (-)-agelastatin total synthesis.
Instead of changing the nucleophilicity of the pyrrole, the electrophilicity of the double bond may be increased by the addition of a Brønsted or Lewis acid. In fact, the biosynthesis of hanishin or longamide B has been described as involving the protonation of a precursor analogous to 7 by an appropriate enzyme [7]. In a bioinspired total synthesis of rac-agelastatin A, a cascade process occurs starting from a hemiaminal 11 that is converted with trifluoroacetic acid (TFA) into a reactive iminium salt 12 that cyclizes to intermediate 13 and then undergoes the addition of water to give the hydroxyl derivative 14. The deprotection of 14 and cyclization by heating in the presence of silica (SiO2) at 45 °C affords agelastatin A (68%) and a minor amount (13%) of its 4,5-epimer [38] (Scheme 3). We can also mention a similar report wherein trifluoroethanol functions as an acidic medium (40 °C) for the diastereoselective cyclization of 14 to agelastatin A [39]. Rac-cyclooroidin has been prepared in excellent yield (93%), by heating the formic acid salt of the acyclic precursor at 95 °C for 45 h in a sealed tube [40].
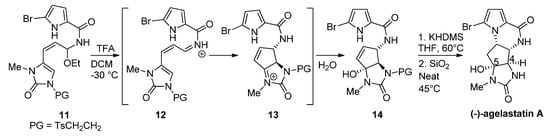
Scheme 3. Silica-promoted synthesis of (-)-agelastatin A.
The palladium-catalyzed cyclization of N-allyl pyrrole-2-carboxamide 15 (R1 = H) leads to different products depending on the catalyst. In the presence of palladium acetate (0.1 eq), sodium acetate and tetrabutylammonium chloride (Bu4NCl) in DMSO at 120 °C, the pyrrolo[1,2-a]pyrazine 16a is formed. On the other hand, PdCl2(CH3CN)2 catalyst (0.1 eq.) in a dimethylformamide (DMF)/tetrahydrofuran(THF) mixture at 100 °C, in the presence of a stoichiometric benzoquinone oxidant, gave a 1:1 mixture of the two isomeric [2,3-c] and [3,2-c] fused pyrrolopyridinone derivatives 17 and 18, apparently as the result of cyclization involving the 2-position of the pyrrole followed by rearrangement [41]. Remarkably, when the Pd(OAc)2 method was applied to the N-cinnamyl derivative 15 (R1 = Ph), the dihydro derivative 19 was obtained in modest yield and different oxidants failed to afford the corresponding pyrrolo[1,2-a]pyrazine 16b [42] (Scheme 4).
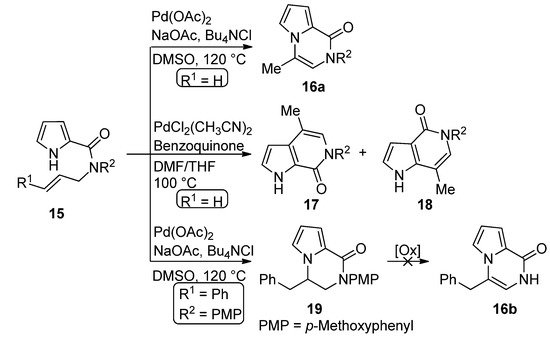
Scheme 4. Palladium-catalyzed cyclization of N-allyl pyrrole-2-carboxamide.
The cyclization reactions of pyrrole nitrogen onto alkyne substituents were studied in basic circumstances. Thus, N-imidazolylpropargyl-substituted pyrrole-2-carboxamide 20 was favorably converted to the pyrrolopyridazinone 21, by an 6-exo-dig process, using cesium carbonate (Cs2CO3) in DMF at room temperature. Further deprotection and exo-double bond reduction yielded cyclooroidin analog 22 [43] (Scheme 5).
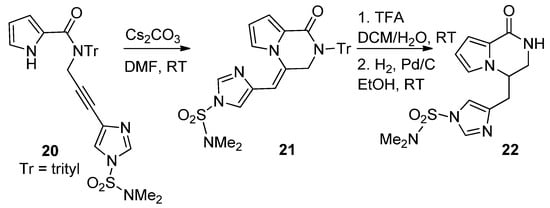
Scheme 5. Base-catalyzed ring closure of N-propargyl pyrrole-carboxamides.
Other examples of the base-catalyzed ring closure of N-propargyl derivatives were reported, with 30 mol% DBU in dichloromethane (DCM) at reflux temperature [44], which led to a mixture of pyrrolopyrazinone isomers 24 and 25 with an exo double bond and an endo double bond, respectively. The isomerization of the exo-isomers 24 to the thermodynamically preferred endo-product 25 and the deprotection mediated by triisopropylsilane (iPr3SiH) and TFA under microwave (MW) heating, only yielded 26 (Scheme 6).
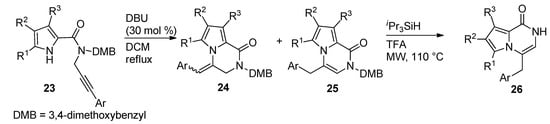
Scheme 6. Ring closure of N-(3-arylpropargyl) pyrrole-carboxamides.
An iridium (I) complex with chiral N-heterocyclic carbene ligand 29 was used as a catalyst for the intramolecular aminoallylation of acylpyrroles 27, leading to (R)-vinyl-substituted pyrrolopyrazinones 28 [45] in e.e. of 92–95% (Scheme 7). In a subsequent report, an Ir/phosphoramidite catalytic system was explored to obtain the (S)-isomer, which is used as a starting material for the total synthesis of longamide B, hanishin or cyclooroidin analogs [46].
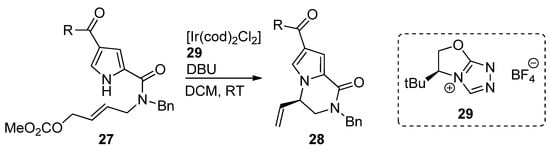
Scheme 7. Iridium-catalyzed intramolecular allylation strategy toward pyrrolopyrazinones.
Pyrrole-2-carboxamides 30 N-substituted with an acetal-protected aldehyde function cyclized upon acid-catalyzed deprotection. The outcome of the reaction is dependent on the reaction conditions. The treatment of 30 with 4-toluenesulfonic acid or HCl in acetone/water at room temperature gave longamide A, probably after the cyclization of the intermediate aldehyde 31. Racemic longamide A can be separated into the two enantiomers through chiral chromatography, but these racemize at room temperature within minutes [47]. On the other hand, the isomeric pyrrolopyridine 32 was formed on the heating of 30 with methanesulfonic acid (MeSO3H). Longamide A was formed on heating with methanesulfonic acid or on treatment with 4-toluenesulfonyl chloride, and trimethylamine gave the dehydrated pyrazinone 33 [48] (Scheme 8). Unprotected ketone analogs of 31 (with different degrees of bromination on the pyrrole ring) were shown to be in equilibrium with the hydroxypyrrolopyrazinones, but the oxidation of the pyrrole ring to a 2-hydroxypyrrolin-5-one with Selectfluor gave the ring-opened product [49].
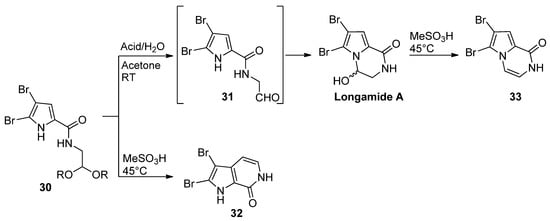
Scheme 8. Alternate intramolecular reactions of acetals and pyrrole.
The pyrrole-2-carbamide 34 (R = H) derived from prolinol on oxidation with 2-iodoxybenzoic acid (IBX) in DMSO at room temperature [50] or Dess–Martin (D-M) reagent in DCM at room temperature [51] gave the hydroxypyrrolopyrazinone 35, which could be dehydrogenated with phosphoryl chloride (POCl3) in pyridine at room temperature [50] or with mesyl chloride and DBU in DCM at room temperature [51] to afford the tricyclic compound 36 (Scheme 9). The compounds 35 and 36 (R = H, Br) were also obtained in a similar sequence from the reduction of the pyrrolecarboxamide connected to the Weinreb amide of proline with lithiumaluminium hydride [52] or with hydroxyprolinate (diisobutylaluminum hydride reduction) [53], in the framework of total syntheses of dibromophakellstatin.
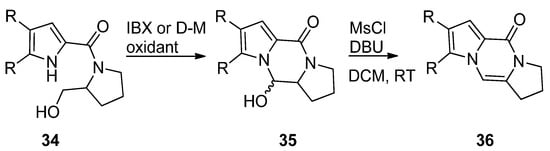
Scheme 9. Cyclization of prolinol derivatives.
Amides 37, resulting from the condensation of 2-trichloroacetylpyrrole (or pyrrole-2-carboxylic acid and amidation reagents) and different amino esters derived from natural amino acids (shown here for proline), were cyclized with sodium hydride in THF to the diketopiperazine derivatives 38 in high yield. Several reports have appeared in the literature [52][54][55][56][57]. These compounds 38 could be oxygenated with molecular oxygen to a hydroperoxide and could be reduced in situ with dibutyl sulfide ((n-Bu)2S) or triphenyl phosphine (PPh3), affording the hydroxy product 39 in high yield [55] (Scheme 10). Recently, it was found that these diketopiperazines 38 could function as catalysts in oxygenation reactions [58], and the oxygenation of compound 38 in the presence of guanidine has also been mentioned as acting in the biogenesis of 2-aminoimidazolidinone metabolites from sponges [54].
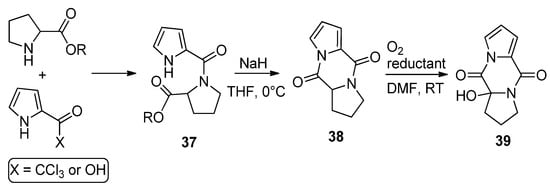
Scheme 10. Diketopiperazine derivatives from amino esters and pyrrole-2-carboxylic reagents.
Pyrrole-2-carboxylic acid, carbonyl compounds, isocyanides and amino esters undergo the four-component Ugi reaction to afford the adducts, which cyclized spontaneously at room temperature in methanol and triethylamine (Et3N) to afford a library of polysubstituted pyrrole diketopyrazines 40 [59] (Scheme 11). An extensive discussion of pyrrolo-fused diketopiperazines is out of the scope of this review. Instead, we give a few additional key references [50][58][60][61][62][63].
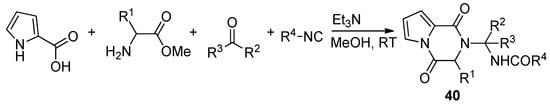
Scheme 11. Pyrrolopyrazinones by Ugi four component reaction.
2.2. Starting from 1-Monosubstituted Pyrroles
There are few examples in the literature of a 1-monosubstitued pyrrole that is converted to a pyrrolopyrazinone. Thus, the pyrrole 41 was prepared from aspartic acid dimethyl ester and reacted with chlorosulfonyl isocyanate (CSI), affording the pyrrolopyrazinedione 42. Reduction with sodium borohydride in methanol, and dehydration, gave the pyrazinone 43, which was then reduced with Pt/C and H2, simultaneously removing the bromine, to longamide B analogs 44 [64] (Scheme 12). Compounds analogous to 42 have also been prepared from the 2-trichloracetylation of 41 followed by substitution with primary amines [20][63].
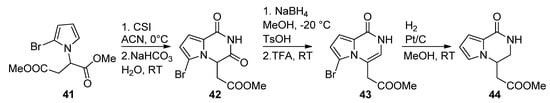
Scheme 12. Cyclization of 1-monosubstituted pyrrole with CSI.
2.3. Starting from 1,2-Disubstituted Pyrroles
There are a number of reports wherein a 1,2-disubstituted pyrrole was used as a starting material for the formation of a pyrrolopyrazinone. This may be done with (1) a single acyclic precursor containing an electrophilic carbonyl group at the 2-position and a nucleophilic substituent (mostly amine) at the 1-position, or (2) vice versa, or (3) the condensation of two components of which one contains the disubstituted pyrrole.
Thus, methyl 2-pyrrolecarboxylate 45 was combined with a nitroalkene 46 in the presence of potassium hydroxide to give a nitroalkyl-substituted pyrrole 47, which was then reduced with sodium borohydride (NaBH4)/cobalt(II)chloride (CoCl2), and the amine 48 cyclized at reflux temperature in toluene after which ethanol was eliminated from 49 in basic medium, leading to the product 50 that was used as a starting material for the first total synthesis of peramide [23][65] (Scheme 13). As an alternative to a nitro compound, a N-CH2CN functionality can be introduced, using iodoacetonitrile, which can be reduced to the amine, which then further cyclizes to a pyrrolopyrazinone [66].
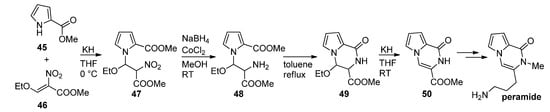
Scheme 13. First total synthesis of peramide.
The azide function is a common precursor for amine that can easily be generated in situ by catalytic reduction. Therefore, in the framework of a total synthesis of cyclooroidin, alcohol 51 was mesylated and converted into azide 52, and catalytic hydrogenation followed by the addition of sodium hydride (NaH) resulted in the formation of the pyrrolopyrazinone 53, which was then further elaborated to cyclooroidin [67] (Scheme 14). Similar strategies have been used in the total synthesis of (-)-hanishin [68] or in the synthesis of histone deacetylase inhibitors [69] and the inhibitors of the mycobacterium ATP synthase [70].
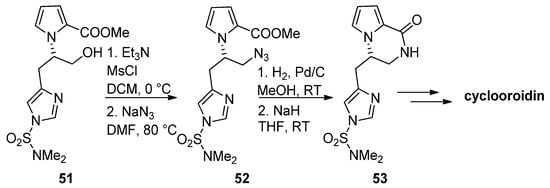
Scheme 14. Azides as intermediates in the total synthesis of cyclooroidin.
Typical amine-protecting groups like tert-butoxycarbonyl (Boc) and fluorenylmethoxycarbonyl (Fmoc) can also be used in intermediates leading to pyrrolopyrazinones. Thus, the condensation of methyl 2-pyrrolocarboxylate 45 with cyclic sulfamidates 54 and the potassium tert-butoxide base gave the precursor 55, which was deprotected with acid and then cyclized, mediated by triethylamine (Et3N) [71]. The resulting pyrrolopyrazinone 56 can then be further elaborated to longamide B or hanishin [72][71][73] (Scheme 15). Further examples of this strategy have been reported toward longamide B derivative, kinase inhibitors [74] and mGluR1 antagonists [66], and we can also mention a Fmoc-based total synthesis of cyclooroidin [75].
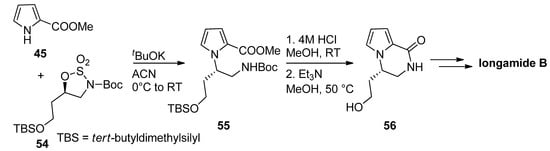
Scheme 15. Boc-strategy toward longamide B derivatives.
An effective example of a two-component reaction leading to the pyrrolopyrazinone scaffold is the reaction of N-(2-bromoethyl)pyrrole-2-carboxylates 57 with amines, leading to the N-substituted bicyclic derivatives 58 (Scheme 16). Probably the reaction starts with the substitution of the bromine by the amine, followed by lactamization. Several examples were reported [76][66][77]. In the framework of agelastatin total synthesis, some examples were reported where, in the presence of sodium hydride, an amide substituted a bromine at the side chain connected to nitrogen, proving that the opposite order of reactions is also possible [78][79].
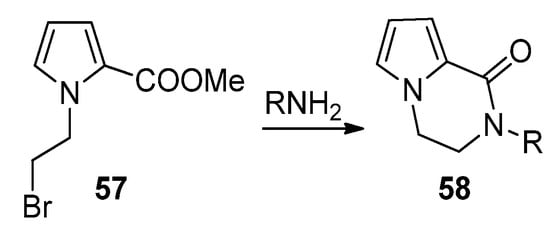
Scheme 16. Bromine substitution and lactamization with primary amines.
The condensation of hydrazine in ethanol with the triester 59 led to the N-aminopyrrolopyrazinone 60, which, upon treatment with sodium nitrite and acid, gave the deaminated derivative 61, and the condensation of 60 with dimethyl acetylenedicarboxylate (DMAD) catalyzed by BF3/acetic acid (BF3·AcOH) complex in acetonitrile afforded the interesting pyrazolo-fused analog 62 [80]. A related cyclization of 1-alkynylpyrrole-2-carboxylate 63 and hydrazine hydrate occurred with remarkable selectivity. Electron-rich aryl groups or alkyl groups R give the pyrrolopyrazinone 64a,b, whereas for R = 4-nitrophenyl, only the 1,2,4-triazine 65c is obtained. The phenyl substituted analogs 63d gave a mixture of the two products 64d and 65d [81] (Scheme 17).
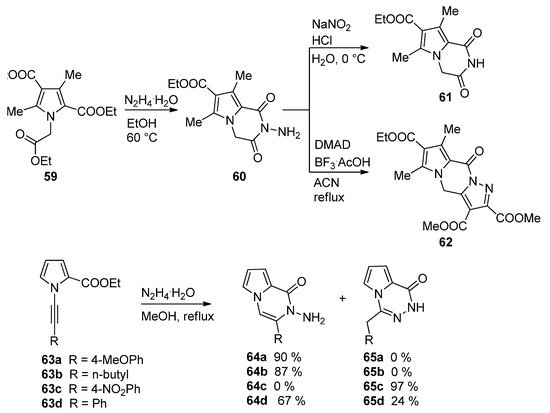
Scheme 17. Reactions of biselectrophilic pyrroles with hydrazine.
N-Propargylpyrrole-2-carboxamides 66 prepared in situ were cyclized to pyrrolopyrazinones 67 using NaH in DMF at room temperature [82], which was applied to a total synthesis of peramide [83] (Scheme 18).
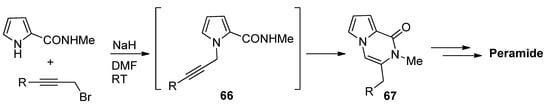
Scheme 18. N-propargylpyrrole-2-carboxamide synthesis and cyclization.
N-(phenacyl)substituted pyrrole-2-carboxylates 68 (R = H) reacted with methylamine (MeNH2) in methanol at reflux to give direct access to pyrrolopyrazinones 69. The diaryl-substituted analogs 68 (R = Ar), on the other hand, gave the amidation product 70, which could be converted to the diaryl analog of 69 (R = Ar) by heating 70 at reflux in a 85% phosphoric acid/ethanol mixture [84] (Scheme 19). An early study of the synthesis of analogs of 69 involved the condensation of amines with intermediate pyrrolo-1,4-oxazines (derived from the N-alkylation of 2-(trichloroacetyl)pyrrole with chloroacetone) [85]. When acetal-protected 1-acetaldehyde 2-carboxamidepyrrole is deprotected in reflux acetic acid, the unsubstituted derivative of pyrrolopyrazinone analog of 69 was obtained [86].
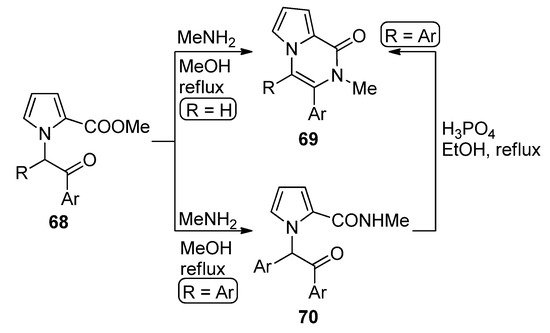
Scheme 19. Cyclization of N-phenacylpyrroles and methylamine.
The carboamination of N-allyl pyrrole carboxamide 71 (R = Ts) with allyl chloride in the presence of 10 mol% Pd(II) hexafluoroacetoacetate (Pd(hfacac)2) and potassium dihydrogenphosphate in toluene/water at 50–80 °C leads to dihydropyrrolopyrazinone 72 [87]. A similar reaction carried out with the same catalyst in the presence of benzoquinone in DMF/water gave the oxygenated analog 73 [88]. Similarly, the carboamination of 71 (R = p-methoxyphenyl, PMP) with aryl bromides in the presence of Pd(OAc)2/S-Phos catalyst at 100 °C afforded the dihydropyrrolopyrazinone 74 [89] (Scheme 20).
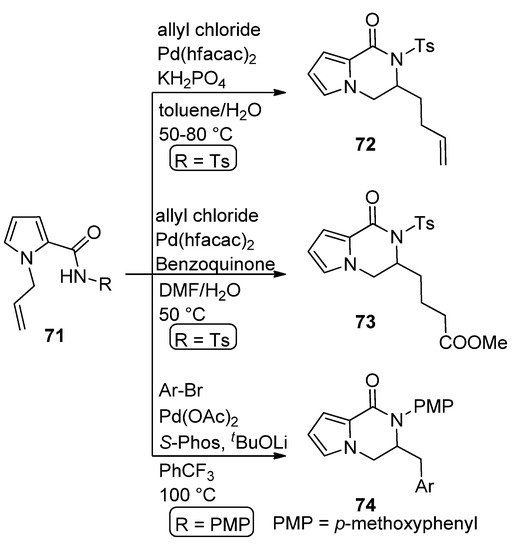
Scheme 20. Pd(II)-catalyzed carboamination reactions.
Allylpalladium species can function as electrophiles in cyclization reactions leading to pyrrolopyrazinones, and this has mainly been used in the context of the total synthesis of natural products. Thus, an enantioselective synthesis of agelastatin A reported by Trost et al. involved firstly the palladium-catalyzed allylation starting from the prochiral bisprotected cyclopentenediol 75 with 5-bromopyrrolecarboxylate 76 in the presence of a chelating chiral bisphosphine catalyst, affording the precursor that then, after conversion to the N-methoxyamide 77, underwent a second intramolecular allylation to afford pyrrolopyrazinone 78, which could then be further elaborated to agelastatin A [90] (Scheme 21). Many variants on this allylation strategy, mostly as a part of agelastatin natural product total syntheses, were reported [91][92][93][94][95][96].
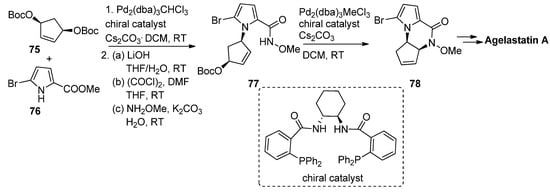
Scheme 21. Palladium-catalyzed intramolecular allylation strategy toward pyrrolopyrazinone.
A remarkable domino reaction of pyrrole-2-carboxamides 79 and vinyl selenones 80 (R2 = H, alkyl) in basic medium occurs via an initial Michael addition, followed by the intramolecular substitution of intermediate 81, leading to pyrrolopyrazinone 82. In the case of styryl selenone 80 (R2 = Ph), the N-(1-phenylethenyl)pyrrole 83 is formed instead [97] (Scheme 22).
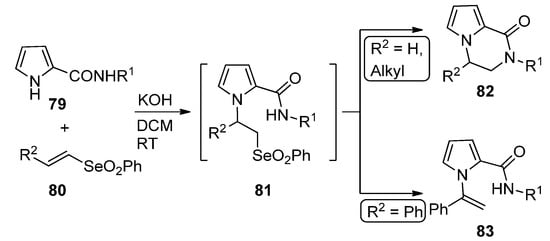
Scheme 22. Domino reaction of pyrrole-2-carboxamides and vinyl selenones.
The Castagnoli–Cushman reaction (CCR) is a ring opening/ring closure reaction of cyclic anhydrides with imines. When applied to anhydride 84, prepared from the diacid with trifluoroacetic anhydride, condensation with different imines 85 in 1,2-dichloroethane (DCE) at room temperature led to a large variety of trisubstituted pyrrolopyrazinones 86 [98] (Scheme 23). The reaction has also been applied to substituted pyrrole anhydrides 84 [99].
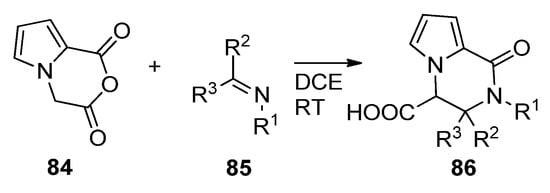
Scheme 23. Castagnoli–Cushman reaction of pyrrole cyclic anhydrides.
The multicomponent Ugi reaction has been applied to N-(2-oxopropyl)pyrrole-2-carboxylic acids 87. In this case, two of the four components (the acid and the ketone) of the Ugi reaction are present on the pyrrole moiety, and two more are added under the form of an isonitrile and an amine. This leads to a library of polysubstituted pyrrolopyrazinones 88 [100] (Scheme 24). Compounds of this type have been described as dengue inhibitors [101].
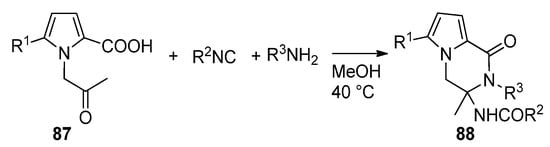
Scheme 24. Ugi reaction toward pyrrolopyrazinones.
Different approaches to rac-dibromophakellin or other tetracyclic marine natural products rely on the intramolecular cycloaddition of a reactive intermediate 90 generated after the oxidation of aminoimidazole 89. This chemistry has been reviewed before [102]. Recently, an intermolecular variant has been described starting from tricyclic 91, which was reacted with guanidine derivative 92 after oxidation with (diacetoxyiodo)benzene (PIDA) and sodium tetrafluoropropoxide (NaTFP) base. Fair amounts of cycloadduct 93 were obtained together with a minor amount of open chain compound 94. The reduction of 93 with an excess of SmI2 then gave the rac-phakellin [103] (Scheme 25). Other approaches involving regio- and stereoselective additions of nitrogen species to analogs of 91 have been mentioned in the framework of dibromophakellstatin total syntheses [51][102][104][105][106].
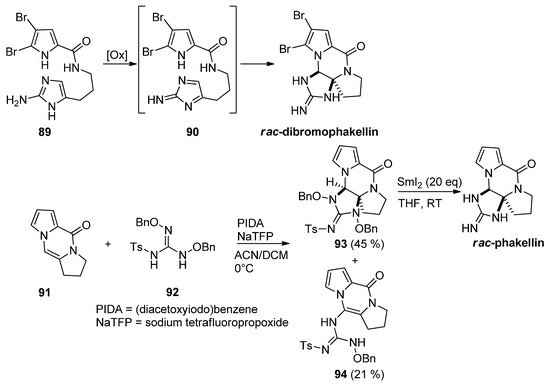
Scheme 25. Oxidative additions of aminoimidazoles and guanidine derivatives toward phakellin natural products.
Different strategies for the total synthesis of palau’amine have been reported, and an exhaustive discussion is beyond the scope of this text, so the reader is referred to some dedicated reviews [107]. One of methods that stand out is the ring contraction of the macrocycle 95 reported by Baran as the final step toward palau’amine [108]. One other remarkable process is a cascade reaction of precursor 96 with the initial deprotonation and ring opening of the tetrahydropyrazole toward intermediate 97a followed by formation or the pyrrolidine ring of intermediate 97b and subsequent formation of the diketopiperazine 98, which then was further elaborated to palau’amine [109] (Scheme 26).
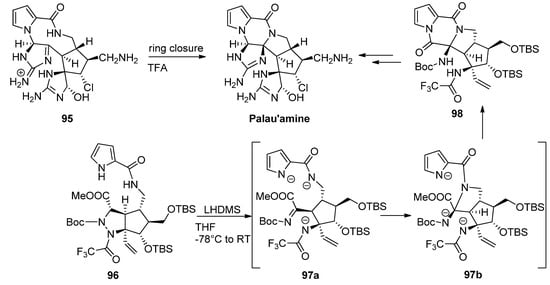
Scheme 26. Different cyclization strategies to palau’amine.
A final strategy toward pyrrolopyrazinones starting from pyrrole building blocks is through the ring expansion of pyrrolizidine derivatives, using the Beckmann rearrangement of the phenyl derivative 99, and, after condensation with hydroxylamine and heating in polyphosphoric acid (PPA), the pyrrolopyrazine 100 is obtained [110] (Scheme 27).
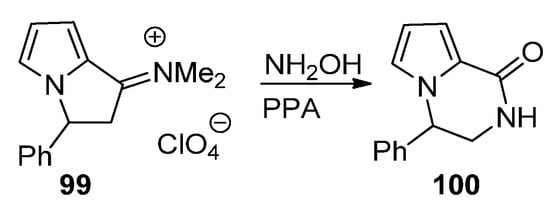
Scheme 27. Ring expansion of pyrrolizidine to pyrrolopyrazinone.
2.4. Fusion of a Pyrrole to a Pyrazinone Derivative
This approach has been much less studied than the pyrrole-first method, with only a few reports so far. Thus, the integrase inhibitors 2 were obtained, starting from pyrazinone 101 and diethyl ethoxymethylene malonate 102, by heating at 100 °C in toluene. The resulting enamine 103 was then cyclized with lithium bis(trimethylsilyl)amide LHMDS at 80 °C in THF, affording compound 2 [111] (Scheme 28).
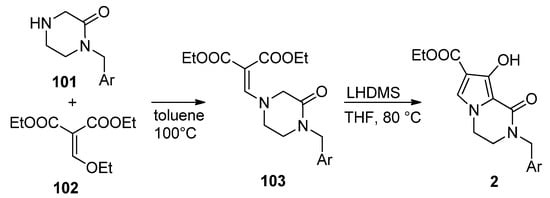
Scheme 28. Synthesis of hydroxy-substituted pyrrolopyrazinones.
An efficient two step synthesis of polysubstituted pyrrolopyrazinones started with the Vilsmeier–Haack chloroformylation of readily available ketones 104 to afford biselectrophilic 2-chloroacrolein intermediate 105, which was then condensed with pyrazinones 106 in the presence of N-methylmorpholine (NMM) base in DMF at 115 °C, affording compounds 107 in fair yields, in some case accompanied with the isomer 108 [112] (Scheme 29).
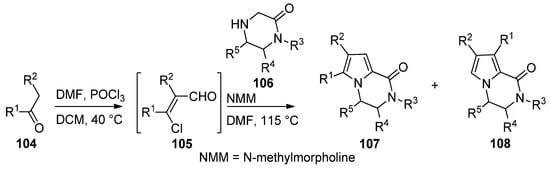
Scheme 29. Two-step synthesis of pyrrolopyrazinones from 2-chloroacroleins.
Isoxazolino-fused piperazinones 111 were prepared via the 1,3-dipolar cycloaddition of nitrones 110, which was in equilibrium with the open chain oximes 109, to dimethyl acetylene dicarboxylate (DMAD). Remarkably, upon heating a rearrangement occurs to pyrrolopyrazinones 112, presumably via a multistep ring contraction/ring expansion mechanism [113] (Scheme 30).
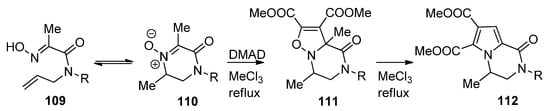
Scheme 30. Synthesis and rearrangement of isoxazolinopyrazinones.
Diketopiperazines 113 underwent base-catalyzed aldol condensations with different aldehydes, affording adducts that, in the case of acetal substituents, as for 114, underwent camphorsulfonic acid (CSA)-mediated cyclization on heating in toluene to pyrrolodiketopiperazines 116. The aldol condensation products 115 resulting from alkynyl aldehydes underwent gold-catalyzed cyclization under similar conditions, giving an alternative preparation for compounds 116 with a larger scope of R1-substituents [114] (Scheme 31).
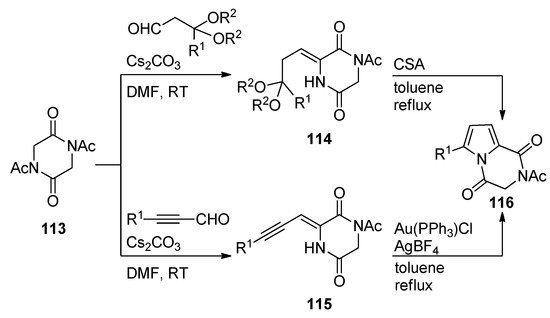
Scheme 31. Brønsted and Lewis acid-catalyzed cyclization reactions of diketopiperazines.
2.5. Multicomponent Reactions
A three-component reaction of 1,2-diaminoethane, dialkyl acetylene dicarboxylate and different biselectrophiles present a very straightforward way to pyrrolopyrazinones. Probably the diamine first reacts with the electrophilic acetylene, and the intermediate pyrazinone derivative 117 then cyclizes with the biselectrophile. Thus, reaction with bromopyruvate in acetonitrile or water at reflux resulted in diester 118 [115][116]. On the other hand, reactions with nitrostyrene, catalyzed by sulfamic acid (SA) in acetonitrile [117] or Fe3O4@SiO2-OSO3H magnetic nanoparticles in water [33] afforded aryl derivatives 119, and condensation with methyl- or arylglyoxal in ethanol at reflux with p-toluenesulfonic acid (TsOH) catalysis gave hydroxyl derivatives 120 [118] (Scheme 32).
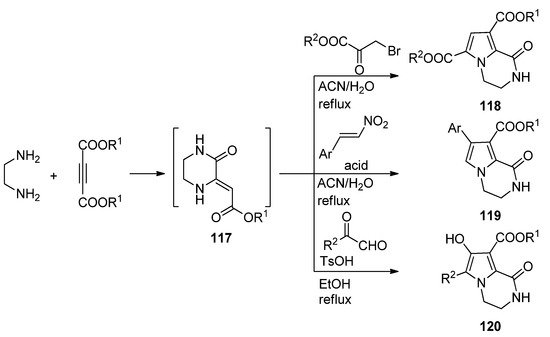
Scheme 32. Three-component reactions leading to dihydropyrrolopyrazinones.
In a variant of this three component reaction, 1,2-diaminoethane and ethyl pyruvate are combined at room temperature in acetonitrile, and then α-bromo ketones are added and the mixture heated in the presence of iron (III) chloride to afford 122 via the reaction of intermediate pyrazine 121 with the bromoketone [119] (Scheme 33).
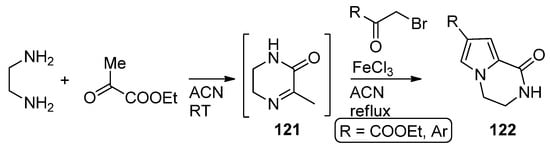
Scheme 33. Three-component reaction with ethyl pyruvate.
2.6. Miscellaneous
The ABDE core of palau’amine was constructed from the dibromide salt of diamine 123 and triscarbonyl compound 124 by a cascade reaction involving a Paal–Knorr pyrrole synthesis, leading to intermediate pyrrole 125, which, after neutralization, undergoes subsequent lactamization to afford tetracyclic 126 [120] (Scheme 34).
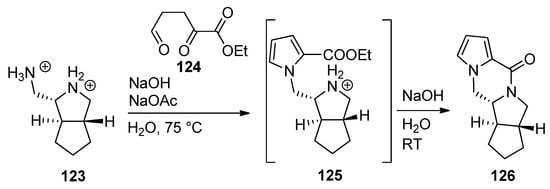
Scheme 34. Cascade process to the ABDE core of palau’amine.
Pyrrolidino-fused diketopiperazines 127 could be oxidized to the pyrrolodiketopiperazines 128 after sequential deprotonation, phenylselenation, oxidation with dimethyldioxirane (DMDO)/elimination and aromatization of the intermediate pyrroline by heating with selenium dioxide in dioxane. Earlier attempts to aromatize 127 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave a lower yield (20–30%) and was accompanied by difficult purification [104]. Recently, the proline-derived compound 129 was oxidized to the pyrrole 130 with MnO2 in THF at 85 °C without effecting the vinyl or dihydropyrazinone parts [121] (Scheme 35).
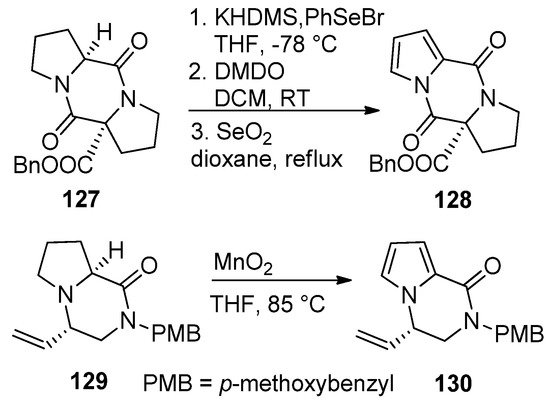
Scheme 35. Aromatization of pyrrolidino-fused diketopiperazines and pyrazinones.
The condensation of N-(2-aminoethyl)aziridine with two equivalents of diethyl acetylenedicarboxylate gave the triester 131, the same compound that could be obtained through the ring transformation of the furan tetraester 132 and 1,2-diaminoethane [122] (Scheme 36).
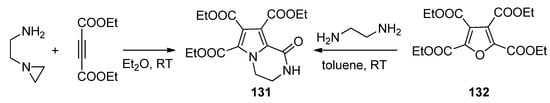
Scheme 36. Formation of pyrrolopyrazinone triesters.
References
- Taylor, A.R.D.; Maccoss, M.; Lawson, A.D.G. Supporting Information Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859.
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274.
- Ritchie, T.J.; Macdonald, S.J.; Young, R.J.; Pickett, S.D. The impact of aromatic ring count on compound developability: Further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov. Today 2011, 16, 164–171.
- Cafieri, F.; Fattorusso, E.; Mangoni, A.; Taglialatela-Scafati, O. Longamide and 3,7-dimethylisoguanine, two novel alkaloids from the marine sponge Agelas longissima. Tetrahedron Lett. 1995, 36, 7893–7896.
- Uemoto, H.; Tsuda, M.; Kobayashi, J. Mukanadins A−C, New Bromopyrrole Alkaloids from Marine SpongeAgelasnakamurai. J. Nat. Prod. 1999, 62, 1581–1583.
- Cafieri, F.; Fattorusso, E.; Taglialatela-Scafati, O. Novel Bromopyrrole Alkaloids from the SpongeAgelas dispar. J. Nat. Prod. 1998, 61, 122–125.
- Mancini, I.; Guella, G.; Amade, P.; Roussakis, C.; Pietra, F. Hanishin, a Semiracemic, Bioactive C9 Alkaloid of the Axinellid Sponge Acanthella carteri from the Hanish Islands. A Shunt Metabolite? Tetrahedron Lett. 1997, 38, 6271–6274.
- Sun, J.; Wu, J.; An, B.; De Voogd, N.J.; Cheng, W.; Lin, W. Bromopyrrole Alkaloids with the Inhibitory Effects against the Biofilm Formation of Gram Negative Bacteria. Mar. Drugs 2018, 16, 9.
- Fattorusso, E.; Taglialatela-Scafati, O. Two novel pyrrole-imidazole alkaloids from the Mediterranean sponge Agelas oroides. Tetrahedron Lett. 2000, 41, 9917–9922.
- Tsuda, M.; Yasuda, T.; Fukushi, E.; Kawabata, J.; Sekiguchi, M.; Fromont, J.; Kobayashi, J. Agesamides A and B, Bromopyrrole Alkaloids from SpongeAgelasSpecies: Application of DOSY for Chemical Screening of New Metabolites. Org. Lett. 2006, 8, 4235–4238.
- Pettit, G.R.; McNulty, J.; Herald, D.L.; Doubek, D.L.; Chapuis, J.-C.; Schmidt, J.M.; Tackett, L.P.; Boyd, M.R. Antineoplastic Agents. 362. Isolation and X-ray Crystal Structure of Dibromophakellstatin from the Indian Ocean SpongePhakellia mauritiana1. J. Nat. Prod. 1997, 60, 180–183.
- D’Ambrosio, M.; Guerriero, A.; Ribes, O.; Pusset, J.; Leroy, S.; Pietra, F. Agelastatin a, a new skeleton cytotoxic alkaloid of the oroidin family. Isolation from the axinellid sponge Agelas dendromorpha of the coral sea. J. Chem. Soc. Chem. Commun. 1993, 1305–1306.
- Hong, T.W.; Jímenez, D.R.; Molinski, T.F. Agelastatins C and D, New Pentacyclic Bromopyrroles from the SpongeCymbastelasp., and Potent Arthropod Toxicity of (−)-Agelastatin A. J. Nat. Prod. 1998, 61, 158–161.
- Kinnel, R.B.; Gehrken, H.P.; Scheuer, P.J. Palau’amine: A cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J. Am. Chem. Soc. 1993, 115, 3376–3377.
- Rowan, D.; Hunt, M.B.; Gaynor, D.L. Peramine, a novel insect feeding deterrent from ryegrass infected with the endophyte Acremonium loliae. J. Chem. Soc. Chem. Commun. 1986, 935–936.
- Jansen, R.; Sood, S.; Mohr, K.I.; Kunze, B.; Irschik, H.; Stadler, M.; Müller, R. Nannozinones and Sorazinones, Unprecedented Pyrazinones from Myxobacteria. J. Nat. Prod. 2014, 77, 2545–2552.
- Song, F.; Liu, X.; Guo, H.; Ren, B.; Chen, C.; Piggott, A.; Yu, K.; Gao, H.; Wang, Q.; Liu, M.; et al. Brevianamides with Antitubercular Potential from a Marine-Derived Isolate of Aspergillus versicolor. Org. Lett. 2012, 14, 4770–4773.
- Trigos, A. Macrophominol, a diketopiperazine from cultures of Macrophomina phaseolina. Phytochemistry 1995, 40, 1697–1698.
- Grube, A.; Köck, M. Oxocyclostylidol, an Intramolecular Cyclized Oroidin Derivative from the Marine Sponge Stylissacaribica§. J. Nat. Prod. 2006, 69, 1212–1214.
- Han, S.; Siegel, D.S.; Morrison, K.C.; Hergenrother, P.J.; Movassaghi, M. Synthesis and Anticancer Activity of All Known (−)-Agelastatin Alkaloids. J. Org. Chem. 2013, 78, 11970–11984.
- Scala, F.; Fattorusso, E.; Menna, M.; Taglialatela-Scafati, O.; Tierney, M.; Kaiser, M.; Tasdemir, D. Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites. Mar. Drugs 2010, 8, 2162–2174.
- Lansdell, T.A.; Hewlett, N.M.; Skoumbourdis, A.P.; Fodor, M.D.; Seiple, I.B.; Su, S.; Baran, P.S.; Feldman, K.S.; Tepe, J.J. Palau’amine and Related Oroidin Alkaloids Dibromophakellin and Dibromophakellstatin Inhibit the Human 20S Proteasome. J. Nat. Prod. 2012, 75, 980–985.
- Brimble, M.A.; Rowanb, D.D. Synthesis of peramine, an insect feeding deterrent mycotoxin from Acremonium lolii. J. Chem. Soc. Chem. Commun. 1988, 4, 978–979.
- Bandini, M.; Eichholzer, A.; Monari, M.; Piccinelli, F.; Umani-Ronchi, A. Versatile Base-Catalyzed Route to Polycyclic Heteroaromatic Compounds by Intramolecular Aza-Michael Addition. Eur. J. Org. Chem. 2007, 2007, 2917–2920.
- Banwell, M.G.; Bray, A.M.; Willis, A.C.; Wong, D.J. First syntheses of the pyrroloketopiperazine marine natural products (±)-longamide, (±)-longamide B, (±)-longamide B methyl ester and (±)-hanishin. New J. Chem. 1999, 23, 687–690.
- Papeo, G.; Frau, M.A.G.-Z.; Borghi, D.; Varasi, M. Total synthesis of (±)-cyclooroidin. Tetrahedron Lett. 2005, 46, 8635–8638.
- Casuscelli, F.; Ardini, E.; Avanzi, N.; Casale, E.; Cervi, G.; D’Anello, M.; Donati, D.; Faiardi, D.; Ferguson, R.D.; Fogliatto, G.; et al. Discovery and optimization of pyrrolo[1,2-a]pyrazinones leads to novel and selective inhibitors of PIM kinases. Bioorg. Med. Chem. 2013, 21, 7364–7380.
- Bandini, M.; Bottoni, A.; Eichholzer, A.; Miscione, G.P.; Stenta, M. Asymmetric Phase-Transfer-Catalyzed Intramolecular N-Alkylation of Indoles and Pyrroles: A Combined Experimental and Theoretical Investigation. Chem. A Eur. J. 2010, 16, 12462–12473.
- Ichikawa, Y.; Yamaoka, T.; Nakano, A.K.; Kotsuki, H. Synthesis of (−)-Agelastatin A by [3.3] Sigmatropic Rearrangement of Allyl Cyanate. Org. Lett. 2007, 9, 2989–2992.
- Davis, F.A.; Deng, J. Asymmetric Total Synthesis of (−)-Agelastatin A Using Sulfinimine (N-Sulfinyl Imine) Derived Methodologies. Org. Lett. 2005, 7, 621–623.
- Domostoj, M.M.; Irving, E.; Scheinmann, F.; Hale, K.J. New Total Synthesis of the Marine Antitumor Alkaloid (−)-Agelastatin A. Org. Lett. 2004, 6, 2615–2618.
- Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. Stereocontrolled Synthesis of Vicinal Diamines by Organocatalytic Asymmetric Mannich Reaction of N-Protected Aminoacetaldehydes: Formal Synthesis of (−)-Agelastatin A. J. Am. Chem. Soc. 2012, 134, 7516–7520.
- Tsuchimochi, I.; Kitamura, Y.; Aoyama, H.; Akai, S.; Nakai, K.; Yoshimitsu, T. Total synthesis of (−)-agelastatin A: An SH2′ radical azidation strategy. Chem. Commun. 2018, 54, 9893–9896.
- Dickson, D.P.; Wardrop, D.J. Total Synthesis of (±)-Agelastatin A, A Potent Inhibitor of Osteopontin-Mediated Neoplastic Transformations. Org. Lett. 2009, 11, 1341–1344.
- Hama, N.; Matsuda, T.; Sato, T.; Chida, N. Total Synthesis of (−)-Agelastatin A: The Application of a Sequential Sigmatropic Rearrangement. Org. Lett. 2009, 11, 2687–2690.
- Feldman, K.S.; Saunders, J.C. Alkynyliodonium Salts in Organic Synthesis. Application to the Total Synthesis of (−)-Agelastatin A and (−)-Agelastatin B. J. Am. Chem. Soc. 2002, 124, 9060–9061.
- Feldman, K.S.; Saunders, J.C.; Wrobleski, M.L. Alkynyliodonium Salts in Organic Synthesis. Development of a Unified Strategy for the Syntheses of (−)-Agelastatin A and (−)-Agelastatin B. J. Org. Chem. 2002, 67, 7096–7109.
- Reyes, J.C.P.; Romo, D. Bioinspired Total Synthesis of Agelastatin A. Angew. Chem. Int. Ed. 2012, 51, 6870–6873.
- Duspara, P.A.; Batey, R.A. A Short Total Synthesis of the Marine Sponge Pyrrole-2-aminoimidazole Alkaloid (±)-Agelastatin A. Angew. Chem. Int. Ed. 2013, 52, 10862–10866.
- Pöverlein, C.; Breckle, G.; Lindel, T. Diels−Alder Reactions of Oroidin and Model Compounds. Org. Lett. 2006, 8, 819–821.
- Beccalli, E.M.; Broggini, G.; Martinelli, M.; Paladino, G.; Maria, B.E. Pd-catalyzed intramolecular cyclization of pyrrolo-2-carboxamides: Regiodivergent routes to pyrrolo-pyrazines and pyrrolo-pyridines. Tetrahedron 2005, 61, 1077–1082.
- Llauger, L.; Bergami, C.; Kinzel, O.D.; Lillini, S.; Pescatore, G.; Torrisi, C.; Jones, P. A novel base-mediated intramolecular hydroamination to build fused heteroaryl pyrazinones. Tetrahedron Lett. 2009, 50, 172–177.
- Bhandari, M.R.; Yousufuddin, M.; Lovely, C.J. Diversity-Oriented Approach to Pyrrole-Imidazole Alkaloid Frameworks. Org. Lett. 2011, 13, 1382–1385.
- Pescatore, G.; Branca, D.; Fiore, F.; Kinzel, O.; Bufi, L.L.; Muraglia, E.; Orvieto, F.; Rowley, M.; Toniatti, C.; Torrisi, C.; et al. Identification and SAR of novel pyrrolo[1,2-a]pyrazin-1(2H)-one derivatives as inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1). Bioorg. Med. Chem. Lett. 2010, 20, 1094–1099.
- Ye, K.-Y.; Cheng, Q.; Zhuo, C.-X.; Dai, L.-X.; You, S. An Iridium(I) N-Heterocyclic Carbene Complex Catalyzes Asymmetric Intramolecular Allylic Amination Reactions. Angew. Chem. Int. Ed. 2016, 55, 8113–8116.
- Zhuo, C.-X.; Zhang, X.; You, S.-L. Enantioselective Synthesis of Pyrrole-Fused Piperazine and Piperazinone Derivatives via Ir-Catalyzed Asymmetric Allylic Amination. ACS Catal. 2016, 6, 5307–5310.
- Marchais, S.; Al Mourabit, A.; Ahond, A.; Poupat, C.; Potier, P. Synthesis of the marine carbinolamine (+/−) longamide control of N-1 and C-3 bromopyrrole nucleophilicity. Tetrahedron Lett. 1999, 40, 5519–5522.
- Sosa, A.C.B.; Yakushijin, K.; Horne, D.A. Controlling cyclizations of 2-pyrrolecarboxamidoacetals. Facile solvation of β-amido aldehydes and revised structure of synthetic homolongamide. Tetrahedron Lett. 2000, 41, 4295–4299.
- Allmann, T.C.; Moldovan, R.-P.; Jones, P.G.; Lindel, T. Synthesis of Hydroxypyrrolone Carboxamides Employing Selectfluor. Chem. A Eur. J. 2015, 22, 111–115.
- Jacquot, D.E.; Hoffmann, H.; Polborn, K.; Lindel, T. Synthesis of the dipyrrolopyrazinone core of dibromophakellstatin and related marine alkaloids. Tetrahedron Lett. 2002, 43, 3699–3702.
- Chung, R.; Yu, E.; Incarvito, C.D.; Austin, D.J. Hypervalent Iodine-Mediated Vicinal Syn Diazidation: Application to the Total Synthesis of (±)-Dibromophakellstatin. Org. Lett. 2004, 6, 3881–3884.
- Travert, N.; Martin, M.-T.; Bourguet-Kondracki, M.-L.; Al-Mourabit, A. Regioselective intramolecular N1–C3 cyclizations on pyrrole–proline to ABC tricycles of dibromophakellin and ugibohlin. Tetrahedron Lett. 2005, 46, 249–252.
- Lindel, T.; Zöllinger, M.; Mayer, P. Enantioselective Total Synthesis of (-)-Dibromophakellstatin. Synlett 2007, 2007, 2756–2758.
- Travert, N.; Al-Mourabit, A. A Likely Biogenetic Gateway Linking 2-Aminoimidazolinone Metabolites of Sponges to Proline: Spontaneous Oxidative Conversion of the Pyrrole-Proline-Guanidine Pseudo-peptide to Dispacamide A. J. Am. Chem. Soc. 2004, 126, 10252–10253.
- Tian, H.; Ermolenko, L.; Gabant, M.; Vergne, C.; Moriou, C.; Retailleau, P.; Al-Mourabit, A. Pyrrole-Assisted and Easy Oxidation of Cyclic α-Amino Acid- Derived Diketopiperazines under Mild Conditions. Adv. Synth. Catal. 2011, 353, 1525–1533.
- Ermolenko, L.; Zhaoyu, H.; Lejeune, C.; Vergne, C.; Ratinaud, C.; Nguyen, T.B.; Al-Mourabit, A. Concise synthesis of Didebromohamacanthin A and Demethylaplysinopsine: Addition of Ethylenediamine and Guanidine Derivatives to the Pyrrole-Amino Acid Diketopiperazines in Oxidative Conditions. Org. Lett. 2014, 16, 872–875.
- Nelli, M.R.; Scheerer, J.R. Synthesis of Peramine, an Anti-insect Defensive Alkaloid Produced by Endophytic Fungi of Cool Season Grasses. J. Nat. Prod. 2016, 79, 1189–1192.
- Petsi, M.; Zografos, A.L. 2,5-Diketopiperazine Catalysts as Activators of Dioxygen in Oxidative Processes. ACS Catal. 2020, 10, 7093–7099.
- Pandey, S.; Khan, S.; Singh, A.; Gauniyal, H.M.; Kumar, B.; Chauhan, P.M.S. Access to Indole- And Pyrrole-Fused Diketopiperazines via Tandem Ugi-4CR/Intramolecular Cyclization and Its Regioselective Ring-Opening by Intermolecular Transamidation. J. Org. Chem. 2012, 77, 10211–10227.
- Imaoka, T.; Akimoto, T.; Iwamoto, O.; Nagasawa, K. Total Synthesis of (+)-Dibromophakellin and (+)-Dibromophakellstatin. Chem. Asian J. 2010, 5, 1810–1816.
- Imaoka, T.; Iwamoto, O.; Nagasawa, K.; Noguchi, K.-I. Total Synthesis of (+)-Dibromophakellin. Angew. Chem. Int. Ed. 2009, 48, 3799–3801.
- Adams, L.A.; Valente, M.W.; Williams, R.M. A concise synthesis of d,l-brevianamide B via a biomimetically-inspired IMDA construction. Tetrahedron 2006, 62, 5195–5200.
- Negoro, T.; Murata, M.; Ueda, S.; Fujitani, B.; Ono, Y.; Kuromiya, A.; Komiya, M.; Suzuki, K.; Matsumoto, J.-I. Novel, Highly Potent Aldose Reductase Inhibitors: (R)-(−)-2-(4-Bromo-2-fluorobenzyl)-1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine- 4-spiro-3‘-pyrrolidine-1,2‘,3,5‘-tetrone (AS-3201) and Its Congeners. J. Med. Chem. 1998, 41, 4118–4129.
- Zhao, D.-G.; Ma, Y.-Y.; Peng, W.; Zhou, A.-Y.; Zhang, Y.; Ding, L.; Du, Z.; Zhang, K. Total synthesis and cytotoxic activities of longamide B, longamide B methyl ester, hanishin, and their analogues. Bioorg. Med. Chem. Lett. 2016, 26, 6–8.
- Brimble, M.A.; Rowan, D.D. Synthesis of the insect feeding deterrent peramine via Michael addition of a pyrrole anion to a nitroalkene. J. Chem. Soc. Perkin Trans. 1 1990, 1, 311–314.
- Micheli, F.; Cavanni, P.; Di Fabio, R.; Marchioro, C.; Donati, D.; Faedo, S.; Maffeis, M.; Sabbatini, F.M.; Tranquillini, M.E. From pyrroles to pyrrolo[1,2-a]pyrazinones: A new class of mGluR1 antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 1342–1345.
- Mukherjee, S.; Sivappa, R.; Yousufuddin, M.; Lovely, C.J. Asymmetric Total Synthesis ofent-Cyclooroidin. Org. Lett. 2010, 12, 4940–4943.
- Das Adhikary, N.; Kwon, S.; Chung, W.-J.; Koo, S. One-Pot Conversion of Carbohydrates into Pyrrole-2-carbaldehydes as Sustainable Platform Chemicals. J. Org. Chem. 2015, 80, 7693–7701.
- Blackburn, C.; Barrett, C.; Brunson, M.; Chin, J.; England, D.; Garcia, K.; Gigstad, K.; Gould, A.; Gutiérrez, J.; Hoar, K.; et al. Histone deacetylase inhibitors derived from 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazine and related heterocycles selective for the HDAC6 isoform. Bioorg. Med. Chem. Lett. 2014, 24, 5450–5454.
- Surase, Y.B.; Samby, K.; Amale, S.R.; Sood, R.; Purnapatre, K.P.; Pareek, P.K.; Das, B.; Nanda, K.; Kumar, S.; Verma, A.K. Identification and synthesis of novel inhibitors of mycobacterium ATP synthase. Bioorg. Med. Chem. Lett. 2017, 27, 3454–3459.
- Fukase, K.; Fujimoto, Y.; Shiokawa, Z.; Inuki, S. Efficient Synthesis of (–)-Hanishin, (–)-Longamide B, and (–)-Longamide B Methyl Ester through Piperazinone Formation from 1,2-Cyclic Sulfamidates. Synlett 2015, 27, 616–620.
- Shiokawa, Z.; Kashiwabara, E.; Yoshidome, D.; Fukase, K.; Inuki, S.; Fujimoto, Y. Discovery of a Novel Scaffold as an Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Based on the Pyrrolopiperazinone Alkaloid, Longamide B. ChemMedChem 2016, 11, 2682–2689.
- Cheng, G.; Wang, X.; Bao, H.; Cheng, C.; Liu, N.; Hu, Y. Total Syntheses of (−)-Hanishin, (−)-Longmide B, and (−)-Longmide B Methyl Ester via a Novel Preparation of N-Substituted Pyrrole-2-Carboxylates. Org. Lett. 2012, 14, 1062–1065.
- Ward, R.A.; Bethel, P.; Cook, C.; Davies, E.; Debreczeni, J.E.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-Guided Discovery of Potent and Selective Inhibitors of ERK1/2 from a Modestly Active and Promiscuous Chemical Start Point. J. Med. Chem. 2017, 60, 3438–3450.
- Mszar, N.W.; Haeffner, F.; Hoveyda, A.H. NHC–Cu-Catalyzed Addition of Propargylboron Reagents to Phosphinoylimines. Enantioselective Synthesis of Trimethylsilyl-Substituted Homoallenylamides and Application to the Synthesis of S-(−)-Cyclooroidin. J. Am. Chem. Soc. 2014, 136, 3362–3365.
- Meyers, K.M.; Méndez-Andino, J.; Colson, A.-O.; Hu, X.E.; Wos, J.A.; Mitchell, M.C.; Hodge, K.; Howard, J.; Paris, J.L.; Dowty, M.; et al. Novel pyrazolopiperazinone- and pyrrolopiperazinone-based MCH-R1 antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 657–661.
- Brimble, M.; Brimble, M.; Hodges, R.; Lane, G. Synthesis of 2-Methylpyrrolo[1,2-a]pyrazin-1(2h)-one. Aust. J. Chem. 1988, 41, 1583–1590.
- Yoshimitsu, T.; Ino, T.; Futamura, N.; Kamon, T.; Tanaka, T. Total Synthesis of the β-Catenin Inhibitor, (−)-Agelastatin A: A Second-Generation Approach Based on Radical Aminobromination. Org. Lett. 2009, 11, 3402–3405.
- Shigeoka, D.; Kamon, T.; Yoshimitsu, T.; Stephenson, C. Formal synthesis of (−)-agelastatin A: An iron(II)-mediated cyclization strategy. Beilstein J. Org. Chem. 2013, 9, 860–865.
- Voievudskyi, M.; Astakhina, V.; Kryshchyk, O.; Petukhova, O.; Shyshkina, S. Synthesis and reactivity of novel pyrrolo[1,2-a]pyrazine derivatives. Mon. Chem. 2015, 147, 783–789.
- Yenice, I.; Basceken, S.; Balci, M. Nucleophilic and electrophilic cyclization of N-alkyne-substituted pyrrole derivatives: Synthesis of pyrrolopyrazinone, pyrrolotriazinone, and pyrrolooxazinone moieties. Beilstein J. Org. Chem. 2017, 13, 825–834.
- Cetinkaya, Y.; Balci, M. Selective synthesis of N-substituted pyrrolo[1,2-a]pyrazin-1(2H)-one derivatives via alkyne cyclization. Tetrahedron Lett. 2014, 55, 6698–6702.
- Ito, S.; Yamamoto, Y.; Nishikawa, T. A concise synthesis of peramine, a metabolite of endophytic fungi. Biosci. Biotechnol. Biochem. 2018, 82, 2053–2058.
- Wang, F.; Wang, J.; Zhang, S. A Novel Synthesis of Arylpyrrolo[1,2-a]pyrazinone Derivatives. Molecules 2004, 9, 574–582.
- Dumas, D.J. Total synthesis of peramine. J. Org. Chem. 1988, 53, 4650–4653.
- Ujjinamatada, R.K. Substituted Tetrahydroquinoline Compounds as ROR Gamma Modulators. WO Patent No. 2016/185342 WIPO, 13 May 2016.
- Hewitt, J.F.M.; Williams, L.; Aggarwal, P.; Smith, C.D.; France, D. Palladium-catalyzed heteroallylation of unactivated alkenes—Synthesis of citalopram. Chem. Sci. 2013, 4, 3538–3543.
- Smith, C.D.; Phillips, D.; Tirla, A.; France, D.J. Catalytic Isohypsic-Redox Sequences for the Rapid Generation of Csp3-Containing Heterocycles. Chem. A Eur. J. 2018, 24, 17201–17204.
- Boothe, J.R.; Shen, Y.; Wolfe, J.P. Synthesis of Substituted γ- and δ-Lactams via Pd-Catalyzed Alkene Carboamination Reactions. J. Org. Chem. 2017, 82, 2777–2786.
- Trost, B.M.; Dong, G. New Class of Nucleophiles for Palladium-Catalyzed Asymmetric Allylic Alkylation. Total Synthesis of Agelastatin A. J. Am. Chem. Soc. 2006, 128, 6054–6055.
- Anderson, G.T.; Chase, C.E.; Koh, Y.-H.; Stien, D.; Weinreb, S.M.; Shang, M. Studies on Total Synthesis of the Cytotoxic Marine Alkaloid Agelastatin A. J. Org. Chem. 1998, 63, 7594–7595.
- Stien, D.; Anderson, G.T.; Chase, C.E.; Koh, Y.-H.; Weinreb, S.M. Total Synthesis of the Antitumor Marine Sponge Alkaloid Agelastatin A. J. Am. Chem. Soc. 1999, 121, 9574–9579.
- Sun, X.-T.; Chen, A. Total synthesis of rac-longamide B. Tetrahedron Lett. 2007, 48, 3459–3461.
- Trost, B.M.; Dong, G. A Stereodivergent Strategy to Both Product Enantiomers from the Same Enantiomer of a Stereoinducing Catalyst: Agelastatin A. Chem. A Eur. J. 2009, 15, 6910–6919.
- Dong, G. Recent advances in the total synthesis of agelastatins. Pure Appl. Chem. 2010, 82, 2231–2246.
- Trost, B.M.; Osipov, M.; Dong, G. Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Vinyl Aziridines with Nitrogen Heterocycles: Rapid Access to Biologically Active Pyrroles and Indoles. J. Am. Chem. Soc. 2010, 132, 15800–15807.
- Palomba, M.; Sancineto, L.; Marini, F.; Santi, C.; Bagnoli, L. A domino approach to pyrazino- indoles and pyrroles using vinyl selenones. Tetrahedron 2018, 74, 7156–7163.
- Chizhova, M.; Khoroshilova, O.; Dar’In, D.; Krasavin, M. Unusually Reactive Cyclic Anhydride Expands the Scope of the Castagnoli–Cushman Reaction. J. Org. Chem. 2018, 83, 12722–12733.
- Chizhova, M.E.; Dar’In, D.V.; Krasavin, M. The Castagnoli–Cushman reaction of bicyclic pyrrole dicarboxylic anhydrides bearing electron-withdrawing substituents. Mendeleev Commun. 2020, 30, 496–497.
- Ilyn, A.P.; Kuzovkova, J.A.; Potapov, V.V.; Shkirando, A.M.; Kovrigin, D.I.; Tkachenko, S.E.; Ivachtchenko, A.V. An efficient synthesis of novel heterocycle-fused derivatives of 1-oxo-1,2,3,4-tetrahydropyrazine using Ugi condensation. Tetrahedron Lett. 2005, 46, 881–884.
- Kounde, C.S.; Yeo, H.-Q.; Wang, Q.-Y.; Wan, K.F.; Dong, H.; Karuna, R.; Dix, I.; Wagner, T.; Zou, B.; Simon, O.; et al. Discovery of 2-oxopiperazine dengue inhibitors by scaffold morphing of a phenotypic high-throughput screening hit. Bioorg. Med. Chem. Lett. 2017, 27, 1385–1389.
- Imaoka, T.; Iwata, M.; Akimoto, T.; Nagasawa, K. Synthetic Approaches to Tetracyclic Pyrrole Imidazole Marine Alkaloids. Nat. Prod. Commun. 2013, 8, 961–964.
- Kovvuri, V.R.R.; Xue, H.; Romo, D. Generation and Reactivity of 2-Amido-1,3-diaminoallyl Cations: Cyclic Guanidine Annulations via Net (3 + 2) and (4 + 3) Cycloadditions. Org. Lett. 2020, 22, 1407–1413.
- Poullennec, K.G.; Romo, D. Enantioselective Total Synthesis of (+)-Dibromophakellstatin. J. Am. Chem. Soc. 2003, 125, 6344–6345.
- Zöllinger, M.; Mayer, P.; Lindel, T. Total Synthesis of the Cytostatic Marine Natural Product Dibromophakellstatin via Three-Component Imidazolidinone Anellation. J. Org. Chem. 2006, 71, 9431–9439.
- Jacquot, D.E.N.; Zöllinger, M.; Lindel, T. Total Synthesis of the Marine Natural Productrac-Dibromophakellstatin. Angew. Chem. Int. Ed. 2005, 44, 2295–2298.
- Ma, Y.; De, S.; Chen, C. Syntheses of cyclic guanidine-containing natural products. Tetrahedron 2015, 71, 1145–1173.
- Seiple, I.B.; Su, S.; Young, I.S.; Lewis, C.A.; Yamaguchi, J.; Baran, P.S. Total Synthesis of Palau’amine. Angew. Chem. Int. Ed. 2009, 49, 1095–1098.
- Namba, K.; Takeuchi, K.; Kaihara, Y.; Oda, M.; Nakayama, A.; Nakayama, A.; Yoshida, M.; Tanino, K. Total synthesis of palau’amine. Nat. Commun. 2015, 6, 8731.
- Robba, M.; Tembo, O.N.; Dallemagne, P.; Rault, S. An Efficient Synthesis of New Phenylpyrrolizine and Phenylpyrrolopyrazine Derivatives. Heterocycles 1993, 36, 2129.
- Fisher, T.E.; Kim, B.; Staas, D.D.; Lyle, T.A.; Young, S.D.; Vacca, J.P.; Zrada, M.M.; Hazuda, D.J.; Felock, P.J.; Schleif, W.A.; et al. 8-Hydroxy-3,4-dihydropyrrolo[1,2-a]pyrazine-1(2H)-one HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6511–6515.
- Sandoval, C.; Lim, N.-K.; Zhang, H. Two-Step Synthesis of 3,4-Dihydropyrrolopyrazinones from Ketones and Piperazin-2-ones. Org. Lett. 2018, 20, 1252–1255.
- Heaney, F.; Fenlon, J.; McArdle, P.; Cunningham, D. α-Keto amides as precursors to heterocycles—generation and cycloaddition reactions of piperazin-5-one nitrones. Org. Biomol. Chem. 2003, 1, 1122–1132.
- Maisto, S.K.; Leersnyder, A.P.; Pudner, G.L.; Scheerer, J.R. Synthesis of Pyrrolopyrazinones by Construction of the Pyrrole Ring onto an Intact Diketopiperazine. J. Org. Chem. 2020, 85, 9264–9271.
- Piltan, M.; Moradi, L.; Abasi, G.; Zarei, S.A.; Wolfe, J.P. A one-pot catalyst-free synthesis of functionalized pyrrolo[1,2-a]quinoxaline derivatives from benzene-1,2-diamine, acetylenedicarboxylates and ethyl bromopyruvate. Beilstein J. Org. Chem. 2013, 9, 510–515.
- Piltan, M. One-Pot, Green and Efficient Synthesis of Pyrrolo[1,2-α]Quinoxalines in Water. J. Chem. Res. 2016, 40, 410–412.
- Moradi, L.; Piltan, M.; Rostami, H.; Abasi, G. One-pot synthesis of pyrrolo[1,2-a]pyrazines via three component reaction of ethylenediamine, acetylenic esters and nitrostyrene derivatives. Chin. Chem. Lett. 2013, 24, 740–742.
- Alizadeh, A.; Abadi, M.H.; Ghanbaripour, R. An Efficient Approach to the Synthesis of Alkyl 7-Hydroxy-1-oxo-1,2,3,4- tetrahydropyrrolo[1,2-a]pyrazine-8-carboxylates via a One-Pot, Three-Component Reaction. Synlett 2014, 25, 1705–1708.
- Piltan, M. One-pot synthesis of pyrrolo[1,2-a]quinoxaline and pyrrolo[1,2-a]pyrazine derivatives via the three-component reaction of 1,2-diamines, ethyl pyruvate and α-bromo ketones. Chin. Chem. Lett. 2014, 25, 1507–1510.
- Aubert-Nicol, S.; Lessard, J.; Spino, C. A Photorearrangement To Construct the ABDE Tetracyclic Core of Palau’amine. Org. Lett. 2018, 20, 2615–2619.
- Jovanovic, M.; Petkovic, M.; Jovanovic, P.; Simic, M.; Tasic, G.; Eric, S.; Savic, V. Proline Derived Bicyclic Derivatives through Metal Catalysed Cyclisations of Allenes: Synthesis of Longamide B, Stylisine D and their Derivatives. Eur. J. Org. Chem. 2019, 2020, 295–305.
- Kostyanovsky, R.G.; El’Natanov, Y.I.; Chervin, I.I.; Antipin, M.Y.; Lyssenko, K.A. Unusual reaction of aziridine dimer with acetylene dicarboxylates. Mendeleev Commun. 1997, 7, 56–58.


