 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shaheed Ur Rehman | + 7411 word(s) | 7411 | 2020-12-08 17:11:03 | | | |
| 2 | Shaheed Ur Rehman | Meta information modification | 7411 | 2020-12-09 18:31:30 | | | | |
| 3 | Shaheed Ur Rehman | Meta information modification | 7411 | 2020-12-09 18:36:21 | | | | |
| 4 | Camila Xu | -3357 word(s) | 4054 | 2020-12-10 08:48:58 | | | | |
| 5 | Camila Xu | -3357 word(s) | 4054 | 2020-12-10 09:01:37 | | |
Video Upload Options
According to WHO report, globally about 10 million active tuberculosis cases, resulting in about 1.6 million deaths, further aggravated by drug-resistant tuberculosis and/or comorbidities. Incomplete therapeutic regimen, meager dosing, and the capability of the latent and/or active state tubercular bacilli to abide and do survive against contemporary first-line and second-line antitubercular drugs escalate the prevalence of drug-resistant tuberculosis. To explore and identify the most potential antitubercular drug candidate among various reported compounds, here we focused to highlight the promising lead derivatives of isoniazid, coumarin, griselimycin, and antimicrobial peptides. The aim of the present review is to fascinate significant lead compounds in the development of potential clinical drug candidates that might be more precise and effective against drug-resistant tuberculosis, the world research looking for a long time.
1. Introduction
Mycobacterium is a genus of Actinobacteria, acid-fast, aerobic, and nonmotile bacteria. This genus contains opportunistic pathogens known to cause serious diseases, especially in the clinical setting. Mycobacterium tuberculosis, the etiological agent of tuberculosis is one of the leading causes of morbidity and mortality in humans [1]. In 80–90% of the cases, these aerobic bacilli enter the lungs, while other extrapulmonary organs, primarily lymph nodes, bones, joints, and the genitourinary system are affected as well [2]. The primary route of transmission of the bacilli into the human lungs is via aerosol droplets [3][4][5].
According to WHO report, globally, about 10 million active tuberculosis cases, resulting in about 1.6 million deaths, further aggravated by drug-resistant tuberculosis and/or comorbidities with diabetes and HIV are present [6]. Drug-resistant tuberculosis is a growing threat to public health. Around half a million new rifampicin-resistant tuberculosis cases (78%—multidrug-resistant tuberculosis) emerged in 2018. Globally, 3.4% of new cases of tuberculosis had multidrug-resistant tuberculosis or rifampicin-resistant tuberculosis (MDR/RR-TB) and 18% of previously treated patients [6][7]. In 2018, an estimated 1.2 million deaths from tuberculosis were reported in HIV-negative persons, while further 251,000 deaths among HIV-positive cases [6] were observed. Tuberculosis co-infection with HIV also raises the risk of relapse per year by up to 10%. Therefore, in patients with HIV coinfection, tuberculosis is the primary cause of death [7].
Antibiotic therapy prolongation contributes to the development of antibiotic resistance. Drug-resistant tuberculosis is an emerging public health threat in many countries of the world. Moreover, the extended duration of antibiotic treatment raises the risk of non-compliance, adverse effects, and drug toxicity. Therefore, it is urgent to find a way to strengthen the current antibiotic therapy regimen against drug-resistant tuberculosis [8]. The process of novel drug development to fight out the resistant strains of tubercular infections has been considered for a long time, and some of the significant candidates are in various stages of clinical trials [9]. Unfortunately, the drug development and approval processes are too lengthy, and these agents are often generally not as effective as expected. In tuberculosis chemotherapy, the other major problem is the relative refractory of mycobacteria to killing by antibiotics or immune systems and the development of resistant strains [10]. Recent advancements in molecular biology and mycobacterial disease immunopathogenesis have contributed to substantial studies in such research areas. Numerous vaccines are manufacturing for prevention or therapeutic purposes [11][12]. We should, however, wait in the coming years for accessible and reliable vaccines. The problematic scenario is the relationship among the host cells and tubercle bacilli afterward the primary infection occurs, which results in latent tuberculosis infection in most cases. As described earlier, the human body can contain the disease in >80% of the cases, and the bacilli remain dormant throughout infected peoples’ entire lives. This phenomenon has been the topic of extensive research [13][14][15]. It is worth mentioning that the WHO, international organizations, educational agencies, foundations, and sponsors are working together to resolve dissimilar facets of the topic and to accomplish the anticipated goals in the approaching periods.
The development of drug-resistant strains of mycobacterium tuberculosis illustrates the importance and demand for early identification of drug-resistant strains, exploring new targets for drug sensitivity, customized treatment plans, and more effective medical interventions. Literature shows several studies, incorporating bioinformatics and proteomics approaches that clearly indicate the potential drug targets and an early diagnostic against drug-resistant strains [16][17][18][19][20]. To tackle the alarming condition of antimicrobial resistance, a pathogen-centric approach covering novel chemotherapeutics and novel diagnostic pathways, along with host targeted therapeutics (i.e., host immune system modulators to treat pathogenesis), must be appraised [21][22][23][24][25]. The efficacy of novel chemotherapeutic agents (i.e., delamanid and bedaquiline), which currently have approval from USFDA, is now compromised by the successional pathogen tolerance strategies [26][27][28][29][30]. Novel antitubercular repurposed drugs as combinational treatment solutions (new anti-TB drug schedules) and host-directed therapeutics may be measured to tackle antibiotic resistance, which is a major problem in tuberculosis management. To combat antibiotic resistance, the key problem in tuberculosis management, we have to identify the most promising lead compounds among new emerging antitubercular agents and conclude these compounds to clinical trials as potential antitubercular drug candidates, along with considering the host-targeted therapeutics.
The use of accelerated molecular microbiological techniques will be the near future of tuberculosis management [12]. Drug tests for susceptibility tests may be either phenotypic or genotypic. Phenotypic tests for drug susceptibility are commonly used with a prime focus on measuring bacterial growth with first-line, e.g., isoniazid, ethambutol (EMB), rifampicin, and streptomycin [SM]), or second-line antitubercular medicines at defined concentrations. This method has certain advantages that it is fast and economical, but has the disadvantage of substantial interruptions and is not reliable for use of ethambutol and other medicine. Genotypical strategies take benefit of particular transmutations relevant to the response against specific medications [13]. Molecular approaches should be conducted using sputum or any other samples and can yield results in a very short period of time (including drug susceptibility). The most used strategies are discussed below:
GenXpert MTB/RIF is an assay for nucleic acid amplification in a sputum sample that analyses DNA and RIF resistance for the presence of mycobacterium tuberculosis. It is a very simple and reproducible procedure with 90% sensitivity and 99% accuracy. The GenXpert is an automated assay and requires no laboratory arrangements. Rifampicin resistance detection is also used for the prediction of MDR-tuberculosis with isoniazid resistance (in most of the cases) [14]. As an initial diagnostic examination, the WHO recently proposed GenXpert for patients with HIV supposed to have tuberculosis or for those who are at risk for rifampicin resistance and/or MDR tuberculosis. GenXpert (MTB/RIF) Ultra is theoretically facilitating, more precise, and sensitive bedside testing that can enhance tuberculosis detection in smear-negative patients, and similar assays are currently under development.
Line probe assay (LPA), approved by WHO, is a family of DNA strip-based tests for swift recognition of first- and second-line antitubercular agents drug resistance [31]. It can also be used for testing culture isolates along with direct testing of acid-fast bacilli as well as smear-positive and -negative sputum specimens. LPA can determine the frequently identified mutations in resistant strains [14][15].
2. Compounds with Promising Antimycobacterial Potentials
Ethambutol, isoniazid, pyrazinamide, rifampicin, amikacin, cycloserine, rifabutin, amoxicillin-clavulanic acid, streptomycin, clofazimine, bedaquiline, aminosalicylic acid, delamanid, ethionamide, moxifloxacin, levofloxacin, linezolid, meropenem, and rifapentine are currently included in the list of essential WHO antituberculosis medications (WHO 2019). The three newly approved bedaquiline, delamanid, and pretomanid showed adverse events and complications when concomitantly used with other antitubercular drugs, and so, they could not be prescribed under routine treatment. Novel antitubercular medicines are less effective, while still a very limited number of the investigational drug compounds are subjected to human clinical trials.
2.1. Classification of Antitubercular Drugs
In the current classification, antitubercular drugs are classified into four groups, i.e., A, B, C, and D (Table 1) [32]. In this classification, it is precisely configured to incorporate the treatment of rifampicin-resistant or MDR-tuberculosis [33].
- Group-A contains fluoroquinolones (high doses of levofloxacin, moxifloxacin, and gatifloxacin). Due to their bactericidal and sterilizing efficacy and strong safety profile, these are called vital products.
- Group-B contains injectable products like (streptomycin, kanamycin, amikacin, and capreomycin) that are incredibly bactericidal but have a lower safety rating than drugs in Group A.
- Ethionamide, cycloserine/terizidone, clofazimine, prothionamide, and linezolid are in Group-C. Given increasing proof of their effectiveness and tolerability, these medications are recommended as vital second-line medicines for multidrug-resistant tuberculosis.
- Group-D is classified into three subgroups: D1: large-dose isoniazid, ethambutol, and pyrazinamide; D2: delamanid and bedaquiline; and D3: para-aminosalicylic acid, meropenem, cilastatin-imipenem, clarithromycin, and clavulanate-amoxicillin.
Some of the emerging novel drugs have the ability to be used as adjuncts in the abovementioned antitubercular regime. The inclusion of such drugs in tuberculosis therapeutics will steer synergistic effects and can lead to improved outcomes by increasing effectiveness against resistant mycobacterium tuberculosis strains (like isoniazid hydrazides and nitazoxanide) or by negating mechanisms of the resistance (like dihydropyridomycin). Furthermore, the development of isoniazid hydrazides and primaquine derivatives enhance the probability of improving patient compliance by decreasing adverse drug reactions and complications.
Table 1: WHO classification of antitubercular drugs.
|
Drug Class |
Included Drugs |
|
(A) Fluoroquinolones |
Levofloxacin, gatifloxacin, moxifloxacin |
|
(B) Second-line injectables |
Streptomycin, kanamycin, amikacin, capreomycin |
|
(C) Other core second-line drugs |
Ethionamide, cycloserine/terizidone, prothionamide, linezolid, clofazimine |
|
(D) Noncore, multidrug-resistant tubercular drugs |
i. High dose—isoniazid, pyrazinamide, ethambutol ii. Delamanid and bedaquiline iii. Para-aminosalicylic acid, meropenem, cilastatin-imipenem, clarithromycin, clavulanate-amoxicillin |
It is promising to see that many potential new candidate compounds being proposed are in pipeline to be significantly used as either monotherapy or combination therapy options to eradicate tuberculosis. However, a variety of prospective drug candidates having direct and/or indirect antitubercular action (just like sildenafil, mefloquine, cilostazol, tizoxanide, metronidazole, entacapone, tolcapone, and ibuprofen) are reported to have high efficacy during in vitro studies as mycobacterial monotherapy or in adjunctive action [31][34]. These compounds need to be tested in vivo and, later on, in human clinical trials. In the enlightenment of the above statement, it is crucial to participate in the recent antitubercular drug discovery campaign that focuses on designing drug regimens capable of shortening treatment duration as well as fight out complicated drug resistance tuberculosis. Thus, it is important to highlight the potential antitubercular drug candidates among others, so, to pursue further research and in vivo studies based on drug safety and efficacy against drug-resistant mycobacterium strains.
2.2. Promising Novel Chemotherapeutics
2.2.1. Isoniazid Lead Derivatives
Isoniazid (isonicotinic hydrazide, pyridine-4-carbohydrazide, and INH) is one of the most significant and efficient first-line antitubercular drugs in tuberculosis therapeutics around the world. Since 1952, isoniazid has been extensively used as a prodrug, which is activated by catalase-peroxidase (KatG), the mycobacterial multifunctional enzyme. The inhibition of mycolic acids synthesis, a distinct building block in the mycobacterium cell wall and accountable for its prominent lipophilic nature, is the main target of isoniazid. The activated isoniazid molecule is responsible for suppressing the biosynthesis of mycolic acid by enoyl-ACP reductase inhibition, an enzyme elaborated in the biosynthesis of fatty acid in the existence of NAD+ or NADH [35]. So, the enoyl-ACP reductase is considered one of the finest-validated targets in the therapeutics of tuberculosis. Isoniazid resistance is mostly mediated by the mutations of the katG gene, which leads to the drug activation incapability, one of the prominent factors responsible for mycobacterium tuberculosis resistance in clinical settings [36]. The mutation rate liable for isoniazid resistance is 100 times greater than rifampicin mutation [37].
Castelo-Branco et al. [38] reported certain isoniazid hydrazide compounds labeled 14, 15, and 16 with striking antimycobacterial potency and decreased hepatotoxicity (lower toxicity to HepG2 cells) as compared to the parent isoniazid compounds. Results showed that mycobacterium strains with both isoniazid KatG (SR 0215, SR 2571, and T113) and rifampicin rpoB (T09) mutations were particularly vulnerable to these isoniazid hydrazides. The MIC for 2-Cyano-N-(4-methylphenyl) acetamide, 2-Chloro-N-(2,6-diethylphenyl) acetamide, and 2-Phenylacetamide were 0.45, 0.43, and 0.47 μM, respectively. Their mycobacterial activity is reliant on the isonicotyl attachment and may be contrary to the isoniazid molecule. In contrast, isoniazid was mutagenic at 50 μM, while its analogs reflected mutagenicity at concentrations >500 μM, under the same assay conditions. Table 2, represents selected chemotherapeutic derivatives and antimicrobial peptides, their molecular targets, mechanism of action, and investigated MIC range for the promising lead compounds.
Table 2: Selected chemotherapeutic derivatives and antimicrobial peptides, their molecular targets, mechanism of action, and investigated minimum inhibitory concentration (MIC) range.
|
Drug Class |
Lead Compounds |
Molecular Targets/Mechanism of Action |
MIC Range |
References |
|
Isoniazid derivatives |
2-Cyano-N-(4-methylphenyl) acetamide, 2-Chloro-N-(2,6-diethylphenyl) acetamide, 2-Phenylacetamide |
Inhibition of mycolic acid synthesis and cell growth inhibitor |
0.43–0.47 μM |
[38,39] |
|
Coumarin derivatives |
6-((3,3-dimethyloxiran-2-yl)-5,7-dihydroxy-8-(2-methylbutanoyl)-4-phenyl-2H-chromen-2-one (1g), Dimethyl substituted compound (1e), coumarin-oxime ether (1h), a coumarin-theophylline hybrid (3a), LSPN270, LSPN271, LSPN476, and LSPN484 |
Cell proliferation inhibitors, cytochrome synthesis disruption, and macrophages activation |
0.12–148 μM |
[41–45] |
|
Griselimycin derivatives |
Cyclohexyl griselimycin |
Inhibits DNA repair and replication |
0.05–0.17 μM |
[46–49] |
|
Antimicrobial peptides |
Bacteriocins (Bcn1–Bcn5), protegrin-1, nisin S, D-V13 K, cathelicidin LL37, D-LAK120-A, and D-LAK120-HP13 |
Multifunctional host immune regulators, pro-inflammatory cytokine responses regulator, calcium influx, and apoptosis |
0.01–30 μM |
[50–56] |
Loots [39] reported that isoniazid was less vulnerable to isoniazid-resistant strains by upregulating the fatty acids and alkanes use and synthesizing bioactive moieties specifically pertain to oxidative stress relief, including those linked to a novel degradation mechanism of ascorbic acid.
Isoniazid, due to its emerging sensitive resistant issues, is going to left out from therapeutic regimens, which endorse the researcher to develop more active novel isoniazid lead derivatives, specifically tailored to target multidrug-resistant tuberculosis. Novel research approaches targeting KatG mutation metabolic schemes and biomarkers have pledged opportunities, which will oblige the discovery of novel isoniazid lead derivatives.
2.2.2. Coumarin Lead Derivatives
The coumarin ring system (chromen-2-one or benzopyran-2-one), existing in natural products that exhibit promising pharmacological properties, has enthralled researchers for decades to probe natural coumarins and their synthetic analogs as potential drug candidates for a wide range of ailments. Coumarins act as intermediate in the synthesis of chromenes, coumarones, furocoumarins, and 2-acylresorcinols [57]. Coumarin and most of its derivatives possess a wide range of therapeutic effects such as antimicrobial [58][59][60][61][62][63], antioxidant [64][65][66], antidepressant [67][68][69], anti-inflammatory [70][71][72][73][74], antitumor [75][76][77][78][79], antinociceptive [80][81][82], antiasthmatic [83][84][85], anti-Alzheimer [85][86][87][88], antipyretic [89][90], and antihyperlipidemic [91][92][93][94] activities. A substantial effort has been made in the past few decades to explore coumarin-based entities as an antitubercular drug, which is agile against clinically approved therapeutic targets reflecting terrific therapeutic outcomes. Over the years, studies on the antimicrobial function of coumarin-based compounds have increased. Coumarin functional nucleus substituted with varied moieties at all positions has resulted in potent antitubercular activity, except for position 1 and 2 [95].
Coumarins have several biological properties that include the inhibition of mycobacterium tuberculosis [96][97][98] and are dependent on structural stability and the scaffolding potential of the parent coumarin compound [45]. Reddy et al. [41] have recently investigated the synthesis of two unique coumarin derivatives, dimethyl substituted compound (1e) and a coumarin-oxime ether derivative (1h), both of which showed excellent antituberculosis activity having MIC of 0.32 and 0.12 μM, respectively. This range is quite comparable to the first-line antitubercular drug isoniazid. According to Mangasuli et al. [42], a coumarin-theophylline hybrid (3a) demonstrated strong binding associations with MTB 4DQU enzymes and potential antimicrobial properties over a broad-spectrum inhibiting Staphylococcus aureus, Escherichia coli, Candida albicans, and Salmonella typhi.
Possible antituberculosis mechanism of action manifested by coumarin derivatives implies cytochrome synthesis disruption, suppression of cell proliferation, macrophage activation, and kinase inhibitor [43]. The insertion of other substituents of interest like heterocyclic moieties and oximes, in the coumarin basic nucleus, could anticipate direction for synthesizing potential novel antitubercular lead compounds.
Natural coumarin 6-((3,3-dimethyloxiran-2-yl)-5,7-dihydroxy-8-(2-methylbutanoyl)-4-phenyl- 2H-chromen-2-one (1g) exhibited the best anti-M. tuberculosis activity with IC50 value of 47.4 μM [41]. Among the synthetic coumarin derivatives, compounds 4-Hydroxy-7-methoxy-2H-chromen-2-one (LSPN270), 7-Ethoxy-4-hydroxy-2H- chromen-2-one (LSPN271), 4-Ethyl-2-hydroxy-4,4a-dihydropyrano[3,2-c]- chromen-5(10bH)-one (LSPN476), and 8-Ethoxy-4-ethyl-2-hydroxy-3,4-dihydropyrano[3,2-c]- chromen-5(2H)-one (LSPN484), which showed MIC from 63.4 μM (≤62.5 μg/mL) against the reference strain and selectivity index between 1.07 and 4.27, demonstrated significant activity against MDR M. tuberculosis clinical isolates (Figure 1) [44].
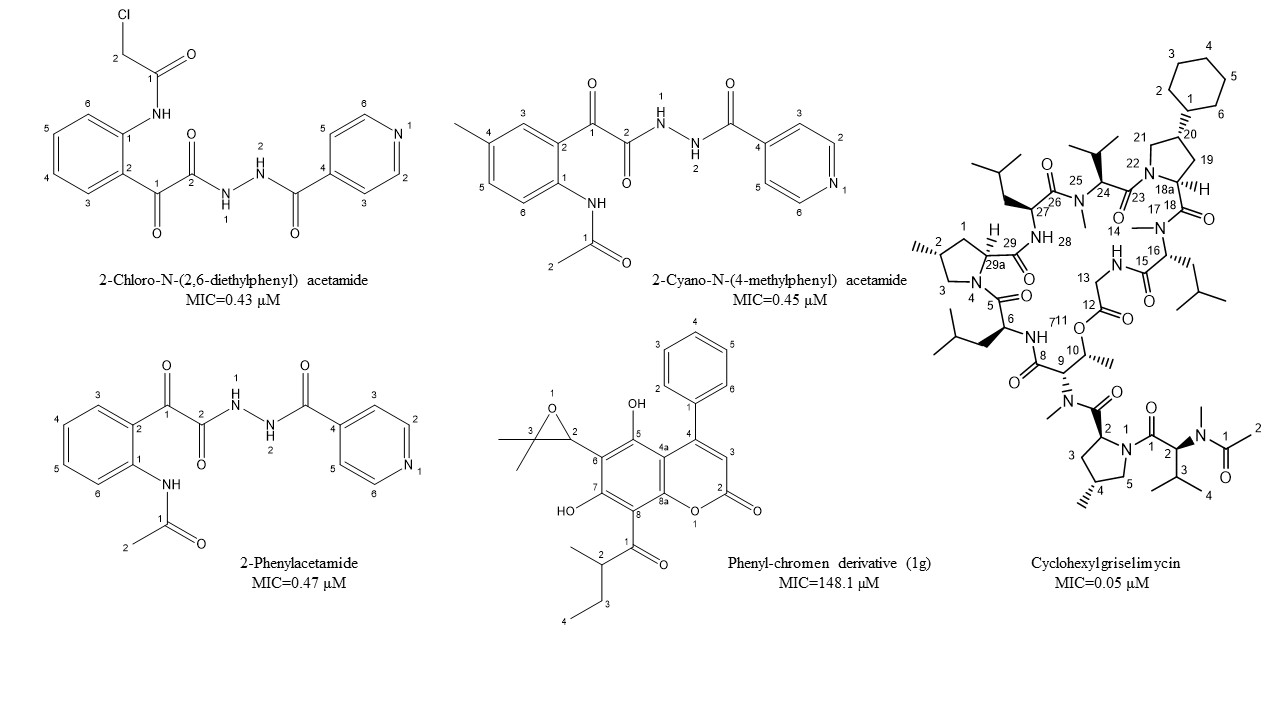
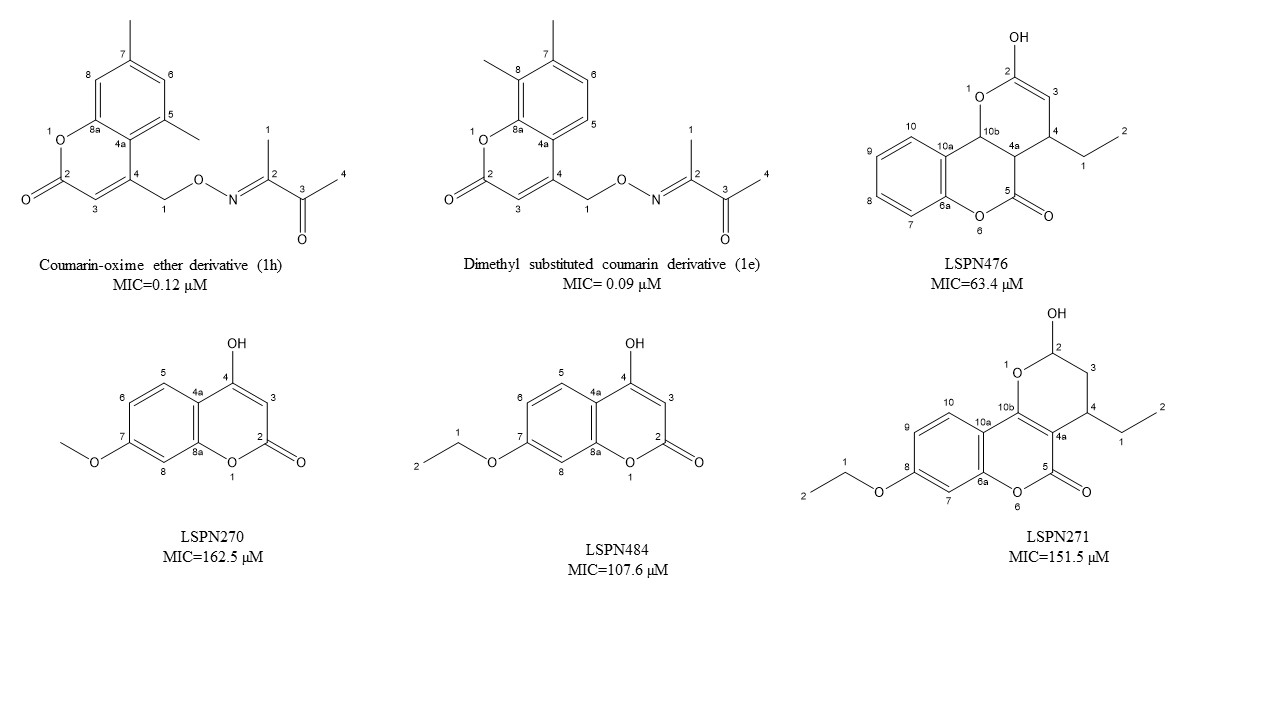
Figure 1. Chemical structure of lead isoniazid, coumarin, and griselimycin derivatives with minimum inhibitory concentrations (MICs) value.
According to Pires et al. [44], compounds 1g, LSPN270, LSPN271, LSPN476, and LSPN484 may be considered potential candidates for further studies in developing new antitubercular drugs.
2.2.3. Griselimycin Lead Derivatives
Griselimycin and its variant methylgriselimycin are natural cyclic compounds synthesized from Streptomyces strain DSM 40835 in the 1960s, which exhibit promising antibacterial and antimycobacterial activity against the resistant strains of M. tuberculosis [99]. Early studies indicated unfavorable pharmacokinetics of griselimycin, given an edge to rifampicin, which was approved at a parallel time frame, while the development of griselimycin as the antimycobacterial drug was break off.
It was determined that the cocrystal structures of griselimycin derivatives attach to DnaN, representing that DnaN peptide-binding pockets have been employed by the cyclic part of the griselimycin antibiotics. The appraisal of the self-resistance to griselimycin in Streptomyces and griselimycin resistance in mycobacterium exhibited amplification of DnaN gene. The DNA polymerase sliding clamp that tie-up the DNA polymerase to DNA and thus confers replicative enzymes responsible for the DNA repair and replication acceleration in prokaryotes [47].
Rolf Müller and his team determined to upgrade griselimycin, considering its promising bioactivity [48]. Primarily, they enhanced the metabolic stability of griselimycin by the addition of alkylation to the proline residue on position 8. Cyclohexyl griselimycin, a synthetic analog of griselimycin, was metabolically stable and showed enhanced lipophilicity and penetration to the thick cell wall of mycobacterium, resulting in superb antimycobacterial activity, with a MIC value of 0.05 μM against the compound susceptible to the mycobacterium tuberculosis H37Rv strain and 0.17 μM for macrophage-like cell encapsulated same strain (RAW264.7) [47][49]. By attacking the DnaN (sliding clamp that anchors the DNA to DNA polymerase), cyclohexyl griselimycins works against mycobacterium tuberculosis and ultimately prevents the proliferation and regeneration of mycobacterial DNA [47][48]. Besides this, significant antimycobacterial activity in acute and chronic tuberculosis mouse models has revealed its synergistic properties with rifampicin and pyrazinamide, with the likelihood of indication for shortening the length of the tuberculosis course [100].
2.3. Lead Antimicrobial Peptides
Antimicrobial peptides (AMPs) also known as host defense peptides are a distinguished part of the innate immune response acquired among all types of life. They are the positively charged peptides, encompass about 12-to-50 amino acid residue [101][102][103]. The existence of the positively charged amino acid moieties, like arginine and lysine, provides a net positive charge to AMPs. The AMPs have hydrophilic and hydrophobic regions, responsible for their amphipathic character, enabling their junction with biological cell membranes while retaining aqueous solubility [32][33]. Following their secondary molecular structure, AMPs are categorized into four categories: α-helix, β-sheet, β-hairpin, and linear non-α-helical loop [104][105]. AMPs principally applied their actions by direct interaction with the microbial cell membrane. Therefore, antibacterial specificity and selectivity of AMPs over the host are quite remarkable. The physiochemical and structural characteristics, including general charge, charge angle, length, conformation, amphipathicity, hydrophobicity, and solubility, exert a considerable effect on the behavior of AMPs [106][107].
Antimicrobial peptides execute an essential role in the generation of an innate immune response, having the modulating capability of the host cellular immunity. AMP expedite pathogen clearance effective for bacteria, viruses, fungi, and parasites, claiming their broad-spectrum antimicrobial coverage. Peptides have been reported to induce the breakdown or alteration of bacterial cell walls and the cytoplasmic elements condensation. AMPs hit the cell wall by various modes of action, like pore formation, thinning, altered curvature, modified electrostatics, and localized perturbations [108]. It has been reported that the cell membrane and cell wall are the primary cathelicidin-based peptide targets, through inserting osmotic action, cell wall synthesis destruction, and likely DNA binding [109].
AMPs are the emerging investigational agents that play a major role in the novel drug development process, and some of them are in Phases-II and -III clinical trials, reflecting their significance and trend in the near future. Interestingly, none of the AMP is in clinical trials for the treatment of tuberculosis. Although many research studies reported significant antimycobacterial activity for AMPs, and even considering the preclinical trials of MU1140 (Oragenics, Inc., Tempa, FL, USA), the essential role of AMPs in tuberculosis therapeutics is clearly indicated.
Several in vitro and in vivo studies revealed significant efficacy of AMPs against drug-resistant strains of mycobacterium. A proline-arginine-rich AMP, PR-39, has proved to be effective against clinical isolates of a multidrug-resistant strain of mycobacterium tuberculosis [110][111]. Fattorini et al. [112] confirmed the inhibitory effect of β-defensin-1 and protegrin-1 against drug-resistant Mycobacterium tuberculosis growth. One other research study investigated the MICs of synthetic peptides from cecropin A and melittin B hybrid against multidrug-resistant strains of tuberculosis [54]. Most of these tested peptides significantly inhibit the growth of multidrug-resistant tuberculosis strains, having MICs comparable to the ones exhibited from susceptible H37Rv strain. AMPs consisting of D-LAK group, having D-amino acids, was also revealed to inhibit multidrug resistance and extended drug resistant strains of tuberculosis, both in vitro and ex vivo [113]. The LL37 (cathelicidin) manifested to have promising efficacy against H37Rv and drug-resistant strains of mycobacterium in the infected mice [53]. Moreover, a 1-month treatment regimen of LL37 (32 μg, three times/week/mouse) indicated for the reduction in up to 53% of a mycobacterial number. It has been denoted that the expression of LL37 is impelled in macrophages due to mycobacterial infection (Figure 2) [114][115].
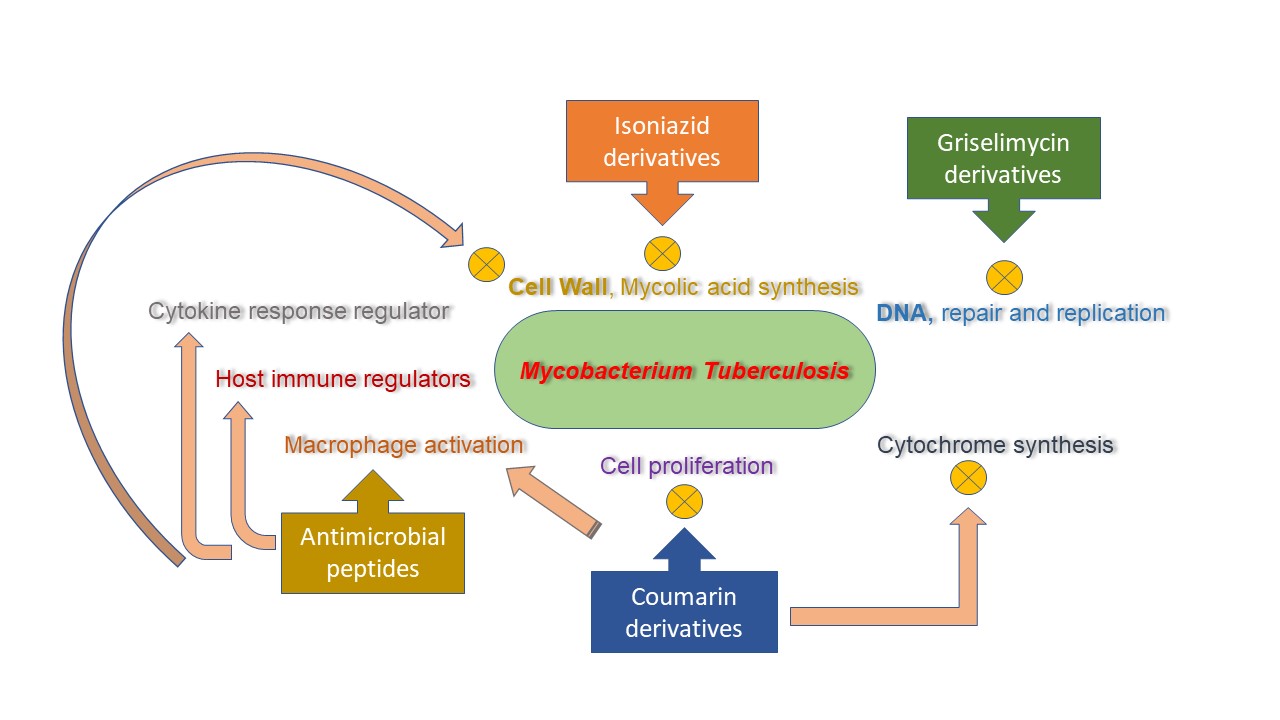
Figure 2. Mechanism of action for isoniazid, coumarin, and griselimycin derivatives and antimicrobial peptides (AMPs).
A 26 amphipathic peptide residue, D-V13 K, comprising all D-amino acids, has positively charged lysine and valine at position no. 16, as “specificity determinant.” It was identified as the most effective analog against mycobacterium tuberculosis having MIC values of 15.6 and 11.2 μM against multidrug-resistant and H37Rv strains, respectively. Peptide D1, having positively charged residue of lysine at the center, demonstrated D5 comparable mycobacterial activity against the multidrug-resistant strains of tuberculosis (i.e., 57 μg/mL) and had a therapeutic index at 7.4-fold higher than D5 [54]. In comparison, D-LAK120-HP13 and D-LAK120-A demonstrated the optimum potency and did not detect mycobacterium in wells at a concentration of 50 μM and above. AMPs’ nonpolar portion has been incorporated into the membrane bilayer and peptide aggregation, resulting in increased permeability, diminish barrier function, cytoplasmic components disintegration, and ultimately cell death [54].
Bacteriocins (Bcns1–Bcn5) group of peptides from different Gram +ev bacteria has been shown to have improved antimycobacterial action against Mycobacterium tuberculosis [55]. Sosunov et al. [55], reported that Bcn4 exhibits excellent bacteriostatic capacity at a concentration of 0.1 μg/mL, which is almost 10-fold lower than the rifampicin MIC50 value. The mechanism of action taken by Bcn1–Bcn5 against mycobacterium tuberculosis is found on the formation of pores in cell membranes, progressing cell death [116][117][118].
The activities of nisin-A and recent biosynthesized hinge derivatives, i.e., nisin-T, nisin-V, and nisin-S, are evaluated for antimycobacterial activity. Both variants indicated an extra antimicrobial activity compared to nisin-A. The nisin-S was reported as the potent one, effective against mycobacterium tuberculosis and mycobacterium avium, eradicating their growth by a rate of 29% relative to nisin-A [56].
Cathelicidins are a large class of mammalian AMPs that are considered an essential part of innate immunity, producing a rapid and nonspecific response to pathogens. By regulating cytokine proinflammatory responses, calcium influx, and apoptosis, cathelicidins are substantially recommended as essential, multifunctional immune regulators with ultimate antimycobacterial activity by the virtue of host adaptive immune responses. The preventive function of cathelicidins in tuberculosis infection depends on the cAMP bursts in macrophages that are activated by mycobacterium, provides tuberculosis pathogenesis clue, and encourages further research into cathelicidin-mediated immune control [52]. NOX2 induces phagocytic activity against intracellular bacilli via the formation of reactive oxygen species (ROS). Yuk et al. [119] reported that NOX2 is necessary for D3-mediated antimycobacterial action via cathelicidin expression.
To the best of our knowledge, studies scrutinizing the antimycobacterial use of AMPs against drug-resistant Mycobacterium tuberculosis have not still got into preclinical trials. However, considering the emerging trend of AMPs and its essential role in the treatment of drug-resistant tuberculosis, it is rational to anticipate such studies in the very near future.
3. Conclusion
Incomplete therapeutic regimen, meager dosing, and the capability of the latent and/or active state tubercular bacilli to abide and to survive against contemporary first-line and second-line antitubercular drugs escalate the prevalence of drug-resistant tuberculosis. It is still a challenge for pharmaceutical scientists to develop the most promising drug substitute among available favorite drug candidates that potentially cover the resistant strain of mycobacterium tuberculosis. In this article, we are fascinated to find the potential drug candidates and their precise tailored derivatives, having promising antimycobacterial properties, but are still on a slow track from a clinical research perspective. Our findings suggested the emerging role and scope of antimicrobial peptides and the lead derivatives of coumarin, isoniazid, and griselimycin having strong antimycobacterial activity in laboratories, which need to accelerate their further in vivo studies. Clinical research on these promising compounds possibly leads to potential antitubercular drug candidates that might be more precise and effective against drug-resistant tuberculosis.
References
- World Health Organization (WHO). WHO Guidelines on Tuberculosis Infection Prevention and Control: 2019 Update; World Health Organization: Geneva, Switzerland,
- Tiemersma, W.; van der Werf, M.J.; Borgdorff, M.W.; Williams, B.G.; Nagelkerke, N.J. Natural history of tuberculosis: Duration and fatality of untreated pulmonary tuberculosis in HIV negative patients: A systematic review. PLoS ONE 2011, 6, e17601.
- Mehta, ; Srivastva, G.; Kachhwaha, S.; Sharma, M.; Kothari, S. Antimycobacterial activity of Citrullus colocynthis (L.) Schrad. against drug sensitive and drug resistant Mycobacterium tuberculosis and MOTT clinical isolates. J. Ethnopharmacol. 2013, 149, 195–200.
- Tascon, ; Soares, C.; Ragno, S.; Stavropoulos, E.; Hirst, E.; Colston, M. Mycobacterium tuberculosis‐activated dendritic cells induce protective immunityin mice. Immunology 2000, 99, 473–480.
- Tian, ; Woodworth, J.; Sköld, M.; Behar, S.M. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J. Immunol. 2005, 175, 3268–3272.
- World Health Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019.
- Harding, WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 2020, 8, 19.
- Dobbs, T.; Webb, R. Chemotherapy of Tuberculosis. Microbiol Spectr, 5 (2). doi: 10.1128/microbiolspec; TNMI7-0040-2017: 2017.
- Shetye, G. S.; Franzblau, S. G.; Cho, S., New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Translational Research 2020, 220, 68-97.
- Young, C.; Walzl, G.; Du Plessis, N., Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal immunology 2019, 1-15.
- Mangtani, ; Abubakar, I.; Ariti, C.; Beynon, R.; Pimpin, L.; Fine, P.E.; Rodrigues, L.C.; Smith, P.G.; Lipman, M.; Whiting, P.F. Protection by BCG vaccine against tuberculosis: A systematic review of randomized controlled trials. Clin. Infect. Dis. 2014, 58, 470–480.
- Kaufmann, H.; Evans, T.G.; Hanekom, W.A. Tuberculosis vaccines: Time for a global strategy. Sci. Transl. Med. 2015, 7, fs8–fs276.
- Wayne, G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163.
- Rustad, R.; Harrell, M.I.; Liao, R.; Sherman, D.R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1502.
- Cardona, ; Ruiz-Manzano, J. On the nature of Mycobacterium tuberculosis-latent bacilli. Eur. Respir. J. 2004, 24, 1044–1051.
- Lata, ; Sharma, D.; Deo, N.; Tiwari, P.K.; Bisht, D.; Venkatesan, K. Proteomic analysis of ofloxacin-mono resistant Mycobacterium tuberculosis isolates. J. Proteom. 2015, 127, 114–121.
- Sharma, ; Bisht, D. Secretory proteome analysis of streptomycin-resistant Mycobacterium tuberculosis clinical isolates. Slas Discov. Adv. Life Sci. R&D 2017, 22, 1229–1238.
- Sharma, ; Bisht, D. M. tuberculosis hypothetical proteins and proteins of unknown function: Hope for exploring novel resistance mechanisms as well as future target of drug resistance. Front. Microbiol. 2017, 8, 465.
- Sharma, ; Bisht, D.; Khan, A.U. Potential alternative strategy against drug resistant tuberculosis: A proteomics prospect. Proteomes 2018, 6, 26.
- Sharma, ; Lata, M.; Singh, R.; Deo, N.; Venkatesan, K.; Bisht, D. Cytosolic proteome profiling of aminoglycosides resistant Mycobacterium tuberculosis clinical isolates using MALDI-TOF/MS. Front. Microbiol. 2016, 7, 1816.
- Lee, ; Lee, J.; Carroll, M.W.; Choi, H.; Min, S.; Song, T.; Via, L.E.; Goldfeder, L.C.; Kang, E.; Jin, B. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 2012, 367, 1508–1518.
- Sotgiu, ; Centis, R.; D’Ambrosio, L.; Spanevello, A.; Migliori, G.B. Linezolid to treat extensively drug-resistant TB: Retrospective data are confirmed by experimental evidence. Eur. Respir. J. 2013, 42, 288–290.
- Dua, ; Rapalli, V.K.; Shukla, S.D.; Singhvi, G.; Shastri, M.D.; Chellappan, D.K.; Satija, S.; Mehta, M.; Gulati, M.; Pinto, T.D.J.A. Multi-drug resistant Mycobacterium tuberculosis & oxidative stress complexity: Emerging need for novel drug delivery approaches. Biomed. Pharmacother. 2018, 107, 1218–1229.
- Alffenaar, ; Van Der Laan, T.; Simons, S.; Van Der Werf, T.; Van De Kasteele, P.; De Neeling, H.; Van Soolingen, D. Susceptibility of clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob. Agents Chemother. 2011, 55, 1287–1289.
- Balasubramanian, ; Solapure, S.; Iyer, H.; Ghosh, A.; Sharma, S.; Kaur, P.; Deepthi, R.; Subbulakshmi, V.; Ramya, V.; Ramachandran, V. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 495–502.
- Andries, ; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 2014, 9, e102135.
- Zimenkov, V.; Nosova, E.Y.; Kulagina, E.V.; Antonova, O.V.; Arslanbaeva, L.R.; Isakova, A.I.; Krylova, L.Y.; Peretokina, I.V.; Makarova, M.V.; Safonova, S.G. Examination of bedaquiline-and linezolid-resistant Mycobacterium tuberculosis isolates from the Moscow region. J. Antimicrob. Chemother. 2017, 72, 1901–1906.
- Veziris, ; Bernard, C.; Guglielmetti, L.; Le Du, D.; Marigot-Outtandy, D.; Jaspard, M.; Caumes, E.; Lerat, I.; Rioux, C.; Yazdanpanah, Y. Rapid emergence of Mycobacterium tuberculosis bedaquiline resistance: Lessons to avoid repeating past errors. Eur. Respir. J. 2017, 49, 3.
- Somoskovi, ; Bruderer, V.; Hömke, R.; Bloemberg, G.V.; Böttger, E.C. A mutation associated with clofazimine and bedaquiline cross-resistance in MDR-TB following bedaquiline treatment. Eur. Respir. J. 2015, 45, 554–557.
- Tadolini, ; Lingtsang, R.D.; Tiberi, S.; Enwerem, M.; D’Ambrosio, L.; Sadutshang, T.D.; Centis, R.; Migliori, G.B. First case of extensively drug-resistant tuberculosis treated with both delamanid and bedaquiline. Eur. Respir. J. 2016, 48, 935–938.
- Harausz, P.; Chervenak, K.A.; Good, C.E.; Jacobs, M.R.; Wallis, R.S.; Sanchez-Felix, M.; Boom, W.H. Activity of nitazoxanide and tizoxanide against Mycobacterium tuberculosis in vitro and in whole blood culture. Tuberculosis 2016, 98, 92–96.
- Hancock, E.; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000, 8, 402–410.
- Ebenhan, ; Gheysens, O.; Kruger, H.G.; Zeevaart, J.R.; Sathekge, M.M. Antimicrobial peptides: Their role as infection-selective tracers for molecular imaging. BioMed Res. Int. 2014, 2014, 867381.
- Vilaplana, ; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J. Infect. Dis. 2013, 208, 199–202.
- Vilchèze, ; Jacobs, J. William R, The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50.
- Machado, ; Perdigao, J.; Ramos, J.; Couto, I.; Portugal, I.; Ritter, C.; Boettger, E.C.; Viveiros, M. High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J. Antimicrob. Chemother. 2013, 68, 1728–1732.
- Petrini, ; Hoffner, S. Drug-resistant and multidrug-resistant tubercle bacilli. Int. J. Antimicrob. Agents 1999, 13, 93–97.
- Castelo-Branco, S.; de Lima, E.C.; de Oliveira Domingos, J.L.; Pinto, A.C.; Lourenço, M.C.S.; Gomes, K.M.; Costa-Lima, M.M.; Araujo-Lima, C.F.; Aiub, C.A.F.; Felzenszwalb, I. New hydrazides derivatives of isoniazid against Mycobacterium tuberculosis: Higher potency and lower hepatocytotoxicity. Eur. J. Med. Chem. 2018, 146, 529–540.
- Loots, T. An altered Mycobacterium tuberculosis metabolome induced by katG mutations resulting in isoniazid resistance. Antimicrob. Agents Chemother. 2014, 58, 2144–2149.
- Rajkhowa, ; C Deka, R. DFT based QSAR/QSPR models in the development of novel anti-tuberculosis drugs targeting Mycobacterium tuberculosis. Curr. Pharm. Des. 2014, 20, 4455–4473.
- Reddy, S.; Kongot, M.; Netalkar, S.P.; Kurjogi, M.M.; Kumar, R.; Avecilla, F.; Kumar, A. Synthesis and evaluation of novel coumarin-oxime ethers as potential anti-tubercular agents: Their DNA cleavage ability and BSA interaction study. Eur. J. Med. Chem. 2018, 150, 864–875.
- Mangasuli, N.; Hosamani, K.M.; Devarajegowda, H.C.; Kurjogi, M.M.; Joshi, S.D. Synthesis of coumarin-theophylline hybrids as a new class of anti-tubercular and anti-microbial agents. Eur. J. Med. Chem. 2018, 146, 747–756.
- Adeniji, A.; Knoll, K.E. Potential anti-TB investigational compounds and drugs with repurposing potential in TB therapy: A conspectus. Appl. Microbiol. Biotechnol. 2020, 104, 5633–5662.
- Pires, T.; Scodro, R.B.; Cortez, D.A.; Brenzan, M.A.; Siqueira, V.L.; Caleffi-Ferracioli, K.R.; Vieira, L.C.; Monteiro, J.L.; Corrêa, A.G.; Cardoso, R.F. Structure–activity relationship of natural and synthetic coumarin derivatives against Mycobacterium tuberculosis. Future Med. Chem. 2020, 12, 1533–1546
- Kapp, ; Visser, H.; Sampson, S.L.; Malan, S.F.; Streicher, E.M.; Foka, G.B.; Warner, D.F.; Omoruyi, S.I.; Enogieru, A.B.; Ekpo, O.E. Versatility of 7-substituted coumarin molecules as antimycobacterial agents, neuronal enzyme inhibitors and neuroprotective agents. Molecules 2017, 22, 1644.
- Dong, ; Pfeiffer, B.; Altmann, K.-H. Recent developments in natural product-based drug discovery for tuberculosis. Drug Discov. Today 2017, 22, 585–591.
- Kling, ; Lukat, P.; Almeida, D.V.; Bauer, A.; Fontaine, E.; Sordello, S.; Zaburannyi, N.; Herrmann, J.; Wenzel, S.C.; König, C. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 2015, 348, 1106–1112.
- Holzgrabe, New griselimycins for treatment of tuberculosis. Chem. Biol. 2015, 22, 981–982.
- Lukat, ; Katsuyama, Y.; Wenzel, S.; Binz, T.; König, C.; Blankenfeldt, W.; Brönstrup, M.; Müller, R. Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem. Sci. 2017, 8, 7521–7527.
- Arranz-Trullén, ; Lu, L.; Pulido, D.; Bhakta, S.; Boix, E. Host antimicrobial peptides: The promise of new treatment strategies against tuberculosis. Front. Immunol. 2017, 8, 1499.
- Lee, -G.; Lee, S.J.; Chae, S.; Lee, K.-Y.; Kim, J.-H.; Lee, B.-J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucl. Acids Res. 2015, 43, 7624–7637.
- Gupta, ; Winglee, K.; Gallo, R.; Bishai, W.R. Bacterial subversion of cAMP signalling inhibits cathelicidin expression, which is required for innate resistance to Mycobacterium tuberculosis. J. Pathol. 2017, 242, 52–61.
- Rivas-Santiago, ; Santiago, C.E.R.; Castañeda-Delgado, J.E.; León–Contreras, J.C.; Hancock, R.E.; Hernandez-Pando, R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2013, 41, 143–148.
- Jiang, ; P Higgins, M.; Whitehurst, J.; O Kisich, K.; I Voskuil, M.; S Hodges, R. Anti-tuberculosis activity of α-helical antimicrobial peptides: De novo designed L-and D-enantiomers versus L-and D-LL37. Protein Pept. Lett. 2011, 18, 241–252.
- Sosunov, ; Mischenko, V.; Eruslanov, B.; Svetoch, E.; Shakina, Y.; Stern, N.; Majorov, K.; Sorokoumova, G.; Selishcheva, A.; Apt, A. Antimycobacterial activity of bacteriocins and their complexes with liposomes. J. Antimicrob. Chemother. 2007, 59, 919–925.
- Carroll, ; Field, D.; O’Connor, P.M.; Cotter, P.D.; Coffey, A.; Hill, C.; O’Mahony, J. The gene encoded antimicrobial peptides, a template for the design of novel anti-mycobacterial drugs. Bioeng. Bugs 2010, 1, 408–412.
- Pereira, M.; Franco, D.P.; Vitorio, F.; Kummerle, A.E. Coumarin compounds in medicinal chemistry: Some important examples from the last years. Curr. Top. Med. Chem. 2018, 18, 124–148.
- Neyts, ; Clercq, E.D.; Singha, R.; Chang, Y.H.; Das, A.R.; Chakraborty, S.K.; Hong, S.C.; Tsay, S.-C.; Hsu, M.-H.; Hwu, J.R. Structure−activity relationship of new anti-Hepatitis C virus agents: Heterobicycle−coumarin conjugates. J. Med. Chem. 2009, 52, 1486–1490.
- Tsay, -C.; Hwu, J.R.; Singha, R.; Huang, W.-C.; Chang, Y.H.; Hsu, M.-H.; Shieh, F.-k.; Lin, C.-C.; Hwang, K.C.; Horng, J.-C. Coumarins hinged directly on benzimidazoles and their ribofuranosides to inhibit hepatitis C virus. Eur. J. Med. Chem. 2013, 63, 290–298.
- Manvar, ; Bavishi, A.; Radadiya, A.; Patel, J.; Vora, V.; Dodia, N.; Rawal, K.; Shah, A. Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37Rv strains. Bioorganic Med. Chem. Lett. 2011, 21, 4728–4731.
- Arshad, ; Osman, H.; Bagley, M.C.; Lam, C.K.; Mohamad, S.; Zahariluddin, A.S.M. Synthesis and antimicrobial properties of some new thiazolyl coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 3788–3794.
- Farshori, N.; Banday, M.R.; Ahmad, A.; Khan, A.U.; Rauf, A. 7-Hydroxy-coumarin derivatives: Synthesis, characterization and preliminary antimicrobial activities. Med. Chem. Res. 2011, 20, 535–541.
- López-Rojas, ; Janeczko, M.; Kubiński, K.; Amesty, Á.; Masłyk, M.; Estévez-Braun, A. Synthesis and antimicrobial activity of 4-substituted 1, 2, 3-triazole-coumarin derivatives. Molecules 2018, 23, 199.
- Kadhum, A.H.; Al-Amiery, A.A.; Musa, A.Y.; Mohamad, A.B. The antioxidant activity of new coumarin derivatives. Int. J. Mol. Sci. 2011, 12, 5747–5761.
- Witaicenis, ; Seito, L.N.; da Silveira Chagas, A.; de Almeida Junior, L.D.; Luchini, A.C.; Rodrigues-Orsi, P.; Cestari, S.H.; Di Stasi, L.C. Antioxidant and intestinal anti-inflammatory effects of plant-derived coumarin derivatives. Phytomedicine 2014, 21, 240–246.
- Manojkumar, ; Ravi, T.; Subbuchettiar, G. Synthesis of coumarin heterocyclic derivatives with antioxidant activity and in vitro cytotoxic activity against tumour cells. Acta Pharm. 2009, 59, 159–170.
- Sashidhara, V.; Kumar, A.; Chatterjee, M.; Rao, K.B.; Singh, S.; Verma, A.K.; Palit, G. Discovery and synthesis of novel 3-phenylcoumarin derivatives as antidepressant agents. Bioorganic Med. Chem. Lett. 2011, 21, 1937–1941.
- Wang, -B.; Liu, H.; Li, G.-Y.; Li, J.; Li, X.-J.; Lei, K.; Wei, L.-C.; Quan, Z.-S.; Wang, X.-K.; Liu, R.-M. Coumarin and 3, 4-dihydroquinolinone derivatives: Synthesis, antidepressant activity, and molecular docking studies. Pharmacol. Rep. 2019, 71, 1244–1252.
- Sashidhara, V.; Rao, K.B.; Singh, S.; Modukuri, R.K.; Teja, G.A.; Chandasana, H.; Shukla, S.; Bhatta, R.S. Synthesis and evaluation of new 3-phenylcoumarin derivatives as potential antidepressant agents. Bioorganic Med. Chem. Lett. 2014, 24, 4876–4880.
- Kontogiorgis, A.; Hadjipavlou-Litina, D.J. Synthesis and antiinflammatory activity of coumarin derivatives. J. Med. Chem. 2005, 48, 6400–6408.
- Fylaktakidou, C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Nicolaides, D.N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004, 10, 3813–3833.
- Bansal, ; Sethi, P.; Bansal, G. Coumarin: A potential nucleus for anti-inflammatory molecules. Med. Chem. Res. 2013, 22, 3049–3060.
- El-Haggar, ; Al-Wabli, R.I. Anti-inflammatory screening and molecular modeling of some novel coumarin derivatives. Molecules 2015, 20, 5374–5391.
- Arora, K.; Kaur, N.; Bansal, Y.; Bansal, G. Novel coumarin–benzimidazole derivatives as antioxidants and safer anti-inflammatory agents. Acta Pharm. Sin. B 2014, 4, 368–375.
- Chen, ; Bi, J.; Su, W. Synthesis and Antitumor Activity of Novel Coumarin Derivatives via a Three‐component Reaction in Water. Chin. J. Chem. 2013, 31, 507–514.
- Qin, -P.; Wang, Z.-F.; Huang, X.-L.; Tan, M.-X.; Zou, B.-Q.; Liang, H. Strong in vitro and vivo cytotoxicity of novel organoplatinum (II) complexes with quinoline-coumarin derivatives. Eur. J. Med. Chem. 2019, 184, 111751.
- Musa, A.; Cooperwood, J.S.; Khan, M.O.F. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr. Med. Chem. 2008, 15, 2664–2679.
- Bisi, ; Cappadone, C.; Rampa, A.; Farruggia, G.; Sargenti, A.; Belluti, F.; Di Martino, R.M.; Malucelli, E.; Meluzzi, A.; Iotti, S. Coumarin derivatives as potential antitumor agents: Growth inhibition, apoptosis induction and multidrug resistance reverting activity. Eur. J. Med. Chem. 2017, 127, 577–585.
- Zhang, ; Li, Z.; Zhou, M.; Wu, F.; Hou, X.; Luo, H.; Liu, H.; Han, X.; Yan, G.; Ding, Z. Synthesis and biological evaluation of 4-(1, 2, 3-triazol-1-yl) coumarin derivatives as potential antitumor agents. Bioorganic Med. Chem. Lett. 2014, 24, 799–807.
- Alipour, ; Khoobi, M.; Emami, S.; Fallah-Benakohal, S.; Ghasemi-Niri, S.F.; Abdollahi, M.; Foroumadi, A.; Shafiee, A. Antinociceptive properties of new coumarin derivatives bearing substituted 3, 4-dihydro-2H-benzothiazines. Daru J. Pharm. Sci. 2014, 22, 9.
- Cheriyan, V., Sr.; Kadhirvelu, P., Sr.; Nadipelly, J., Jr.; Shanmugasundaram, J.; Sayeli, V., Sr.; Subramanian, V., Sr. Anti-nociceptive effect of 7-methoxy coumarin from Eupatorium Triplinerve vahl (Asteraceae). Pharmacogn. Mag. 2017, 13, 81.
- Park, -H.; Sim, Y.-B.; Kang, Y.-J.; Kim, S.-S.; Kim, C.-H.; Kim, S.-J.; Lim, S.-M.; Suh, H.-W. Antinociceptive profiles and mechanisms of orally administered coumarin in mice. Biol. Pharm. Bull. 2013, 36, 925–930.
- Sanchez-Recillas, ; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Rios, M.Y.; Ibarra-Barajas, M.; Estrada-Soto, S. Semisynthesis, ex vivo evaluation, and SAR studies of coumarin derivatives as potential antiasthmatic drugs. Eur. J. Med. Chem. 2014, 77, 400–408.
- Leal, K.A.M.; Silva, A.H.; de Barros Viana, G.S. Justicia pectoralis, a coumarin medicinal plant have potential for the development of antiasthmatic drugs? Rev. Bras. Farmacogn. 2017, 27, 794–802.
- Wang, ; Fu, Y.; Wei, Z.; He, X.; Shi, M.; Kou, J.; Zhou, E.; Liu, W.; Yang, Z.; Guo, C. Anti-asthmatic activity of osthole in an ovalbumin-induced asthma murine model. Respir. Physiol. Neurobiol. 2017, 239, 64–69.
- Piazzi, ; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorganic Med. Chem. Lett. 2008, 18, 423–426.
- Shaik, B.; Palaka, B.K.; Penumala, M.; Kotapati, K.V.; Devineni, S.R.; Eadlapalli, S.; Darla, M.M.; Ampasala, D.R.; Vadde, R.; Amooru, G.D. Synthesis, pharmacological assessment, molecular modeling and in silico studies of fused tricyclic coumarin derivatives as a new family of multifunctional anti-Alzheimer agents. Eur. J. Med. Chem. 2016, 107, 219–232.
- Patil, O.; Bari, S.B.; Firke, S.D.; Deshmukh, P.K.; Donda, S.T.; Patil, D.A. A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorganic Med. Chem. 2013, 21, 2434–2450.
- Ahmad, ; Asad, M.; Siddiqui, Z.N. Evaluation of antipyretic and antinociceptive potential of new heterocyclic derivatives of 3-formyl-4-hydroxycoumarin in rats. Int. Res. J. Pharm. Appl. Sci. 2013, 3, 253–259.
- El-Sharkawy, A.; AlBratty, M.M.; Alhazmi, H.A. Synthesis of some novel pyrimidine, thiophene, coumarin, pyridine and pyrrole derivatives and their biological evaluation as analgesic, antipyretic and anti-inflammatory agents. Braz. J. Pharm. Sci. 2018. https://doi.org/10.1590/s2175-97902018000400153
- Sashidhara, V.; Kumar, A.; Kumar, M.; Sonkar, R.; Bhatia, G.; Khanna, A. Novel coumarin derivatives as potential antidyslipidemic agents. Bioorganic Med. Chem. Lett. 2010, 20, 4248–4251.
- Sashidhara, V.; Kumar, A.; Kumar, M.; Srivastava, A.; Puri, A. Synthesis and antihyperlipidemic activity of novel coumarin bisindole derivatives. Bioorganic Med. Chem. Lett. 2010, 20, 6504–6507.
- Asif, Pharmacologically potentials of different substituted coumarin derivatives. Chem. Int. 2015, 1, 1–11.
- Pari, ; Rajarajeswari, N.; Saravanan, S.; Rathinam, A. Antihyperlipidemic effect of coumarin in experimental type 2 diabetic rats. Biomed. Prev. Nutr. 2014, 4, 171–176.
- Keri, S.; Sasidhar, B.; Nagaraja, B.M.; Santos, M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015, 100, 257–269.
- Yang, ; Zhao, N.; Song, J.; Zhu, K.; Jiang, C.-s.; Shan, P.; Zhang, H. Design, synthesis and biological evaluation of novel coumarin-based hydroxamate derivatives as histone deacetylase (hdac) inhibitors with antitumor activities. Molecules 2019, 24, 2569.
- Abdizadeh, ; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Ghodsi, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62.
- Niu, ; Wang, W.; Li, J.; Lei, Y.; Zhao, Y.; Yang, W.; Zhao, C.; Lin, B.; Song, S.; Wang, S. A novel structural class of coumarin-chalcone fibrates as PPARα/γ agonists with potent antioxidant activities: Design, synthesis, biological evaluation and molecular docking studies. Eur. J. Med. Chem. 2017, 138, 212–220.
- Terlain, ; Thomas, J. Structure of griselimycin, polypeptide antibiotic extracted from streptomyces cultures. II. Structure of griselimycin. Bull. Soc. Chim. Fr. 1971, 6, 2357.
- Herrmann, ; Rybniker, J.; Müller, R. Novel and revisited approaches in antituberculosis drug discovery. Curr. Opin. Biotechnol. 2017, 48, 94–101.
- Brogden, A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250.
- Nguyen, T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472.
- Fjell, D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51.
- Giuliani, ; Pirri, G.; Nicoletto, S. Antimicrobial peptides: An overview of a promising class of therapeutics. Open Life Sci. 2007, 2, 1–33.
- Bahar, A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575.
- Pushpanathan, ; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013, 675391.
- Jindal, ; Le, C.; Mohd Yusof, M.; Sekaran, S. Net charge, hydrophobicity and specific amino acids contribute to the activity of antimicrobial peptides. J. Health Transl. Med. 2014, 17, 1–7.
- Abedinzadeh, ; Gaeini, M.; Sardari, S. Natural antimicrobial peptides against Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2015, 70, 1285–1289.
- Kang, -J.; Kim, D.-H.; Mishig-Ochir, T.; Lee, B.-J. Antimicrobial peptides: Their physicochemical properties and therapeutic application. Arch. Pharmacal Res. 2012, 35, 409–413.
- Linde, M.; Hoffner, S.E.; Refai, E.; Andersson, M. In vitro activity of PR-39, a proline-arginine-rich peptide, against susceptible and multi-drug-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2001, 47, 575–580.
- Hao, ; Shi, Y.-H.; Tang, Y.-L.; Le, G.-W. The intracellular mechanism of action on Escherichia coli of BF2-A/C, two analogues of the antimicrobial peptide Buforin 2. J. Microbiol. 2013, 51, 200–206.
- Fattorini, ; Gennaro, R.; Zanetti, M.; Tan, D.; Brunori, L.; Giannoni, F.; Pardini, M.; Orefici, G. In vitro activity of protegrin-1 and beta-defensin-1, alone and in combination with isoniazid, against Mycobacterium tuberculosis. Peptides 2004, 25, 1075–1077.
- Lan, ; Lam, J.T.; Siu, G.K.; Yam, W.C.; Mason, A.J.; Lam, J.K. Cationic amphipathic D-enantiomeric antimicrobial peptides with in vitro and ex vivo activity against drug-resistant Mycobacterium tuberculosis. Tuberculosis 2014, 94, 678–689.
- Rivas-Santiago, ; Hernandez-Pando, R.; Carranza, C.; Juarez, E.; Contreras, J.L.; Aguilar-Leon, D.; Torres, M.; Sada, E. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect. Immun. 2008, 76, 935–941.
- Santos, C.; Silva‐Gomes, S.; Silva, J.P.; Gama, M.; Rosa, G.; Gallo, R.L.; Appelberg, R. Endogenous cathelicidin production limits inflammation and protective immunity to Mycobacterium avium in mice. Immun. Inflamm. Dis. 2014, 2, 1–12.
- Chatterjee, ; Paul, M.; Xie, L.; Van Der Donk, W.A. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 2005, 105, 633–684.
- Guinane, ; Cotter, P.; Hill, C.; Ross, R. Microbial solutions to microbial problems; lactococcal bacteriocins for the control of undesirable biota in food. J. Appl. Microbiol. 2005, 98, 1316–1325.
- Carroll, ; Draper, L.A.; O’Connor, P.M.; Coffey, A.; Hill, C.; Ross, R.P.; Cotter, P.D.; O’Mahony, J. Comparison of the activities of the lantibiotics nisin and lacticin 3147 against clinically significant mycobacteria. Int. J. Antimicrob. Agents 2010, 36, 132–136.
- Yuk, -M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243.


