Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Texali Garcia | + 5023 word(s) | 5023 | 2021-07-05 09:00:40 | | | |
| 2 | Conner Chen | -1205 word(s) | 3818 | 2021-08-16 05:44:53 | | | | |
| 3 | Conner Chen | -1205 word(s) | 3818 | 2021-08-16 05:45:45 | | | | |
| 4 | Conner Chen | -1205 word(s) | 3818 | 2021-08-16 05:46:47 | | | | |
| 5 | Conner Chen | Meta information modification | 3818 | 2021-08-16 12:15:17 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Garcia, T.; Cambrón-Mora, D. RAAS in HF and Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/13175 (accessed on 07 February 2026).
Garcia T, Cambrón-Mora D. RAAS in HF and Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/13175. Accessed February 07, 2026.
Garcia, Texali, Diego Cambrón-Mora. "RAAS in HF and Cancer" Encyclopedia, https://encyclopedia.pub/entry/13175 (accessed February 07, 2026).
Garcia, T., & Cambrón-Mora, D. (2021, August 14). RAAS in HF and Cancer. In Encyclopedia. https://encyclopedia.pub/entry/13175
Garcia, Texali and Diego Cambrón-Mora. "RAAS in HF and Cancer." Encyclopedia. Web. 14 August, 2021.
Copy Citation
Heart failure (HF) and cancer are the main public health issues in industrialized countries and are increasing in prevalence, especially, in the ageing population. These two diseases were thought to be independent; however, new research has revealed that cancer and HF frequently coexist in the same patient. Furthermore, as cancer-specific mortality decreases and the surviving population gets older, the overlap between cardiac disease and cancer patients is growing. As a result, the discipline of cardio-oncology has primarily focused on the adverse effects of anti-cancer therapy. HF is one of the most serious consequences of cardiotoxic cancer therapy.
heart failure
myocardial infarction
cancer
renin-angiotensin-aldosterone system
1. Renin-Angiotensin-Aldosterone System Linking Ischemic Heart Failure and Cancer
There is evidence that the RAAS is involved in most of the tumorigenic characteristics described above, and due to the chronic activation of RAAS in HF, it has been proposed that a failing heart may be closely related to the development of cancer. In this review, we will discuss recent studies that highlight the role of RAAS components as an axis of crucial importance in the pathophysiology of HF and as well as evidence of the dysregulation of its components in the development of cancer to highlight the points where these two entities that were previously considered independent could now converge.
There are two major pathways in the RAAS: classical and non-classical pathways. In the classical RAAS, the effector peptide is angiotensin-II (AngII), which is produced from its hepatic precursor, angiotensinogen, which is catabolized by the enzyme renin, giving rise to angiotensin-I (AngI) in turn, which is a substrate for the angiotensin-converting enzyme (ACE) producing angiotensin-II. The functional effects of AngII in the classical RAAS are largely mediated by the type 1 angiotensin-II receptor (AT1R) and the type 2 receptor (AT2R) [1][2]. AT1R activation increases aldosterone, an important player in the regulation of electrolyte balance [3], but AT1R activation also has many other effects (described later). Signaling mediated by the AT2R is associated with antifibrotic functions and even with anti-inflammatory effects in HF [4][5][6], while in cancer, this axis has antiproliferative, antiangiogenic, and pro-apoptotic effects [7][8]. However, there are also conflicting reports suggesting possible tumor type-specific differences [3][7].
In the non-classical RAAS, the homologue of ACE, angiotensin-converting enzyme 2 (ACE2) cleaves AngI into a nonapeptide, Ang 1-9 and AngII into a heptapeptide, Ang 1-7. Additionally, AngII can be also converted to Ang 2-8 (AngIII) by aminopeptidase A, and exerts its effects by binding to AT1R. Aminopeptidase N converts AngIII to Ang 3-8 (AngIV) and can act through the angiotensin 4 receptor (AT4R) [1]. Ang 1-9 can activate AT2R, and Ang 1-7 can bind to the proto-oncogene Mas receptor (MasR). Interestingly, every one of these components has been demonstrated to counteract the actions of the classical RAAS [1][9]. Signaling mediated by the ACE2/Ang 1-7/MasR axis has been shown to have a protective role in the development of myocardial remodeling post-MI in an animal model [10], but it is also associated with antifibrotic and anti-inflammatory effects [11][12]. Moreover, AngIV/AT4R signaling has a cardioprotective role, acting as a counterpart of Ang II-mediated inflammation and myocardial fibrosis in rat model [13]. In cancer, MasR has been documented to reduce abnormal angiogenesis, inflammation and cell proliferation by the local decrease of Ang II levels or AT1 receptor blockade associated with high concentrations of Ang(1-7) at the tumor site [14]. Even so, as the AT1R continues to be crucial in mediating physiological and pathophysiological effects of AngII [15], in this review, we are going to focus in the classical AT1R/AngII RAAS axis.
AngII overproduction is linked to the development of chronic illnesses; in fact, a chronic activation of RAAS is a hallmark of HF, especially marked by a systemic increase in levels of AngII [16][17], and to better understand how RAAS is implied in both of these diseases, first we must consider the involved pathophysiology from MI to HF and then later to cancer.
Cardiomyocyte necrosis in the infarcted myocardium activates the innate immune response, triggering an inflammatory response. The release of danger signals from dying cells induces the secretion of cytokines, like chemokines and adhesion molecules, to allow the recruitment and infiltration of leukocytes, mainly monocytes, into the infarcted area, where they exert a “reparative” response, phagocytosing the cellular debris, while stimulating repair pathways by secreting pro-inflammatory cytokines. To supply the appropriate number of immune cells, a release of stem cells and hematopoietic progenitors from the niches of the bone marrow occurs; these cells then migrate to the spleen and, ultimately, increase the production of immune cells, which in turn mediates an effective inflammatory response (Figure 1) [18][19]. The modulation of inflammation in this repair phase includes fibroblast activation and healing mediated by the neurohumoral response. RAAS, which is part of the neurohumoral response, is activated by renal hypoperfusion and sympathetic activation as compensatory mechanisms after a myocardial injury [20]. In these processes, RAAS actively participates mainly through the AngII effector peptide. Indeed, various components of RAAS, including angiotensinogen, AngII, ACE, AT1R, and AT2R, have been reported to be expressed in a variety of immune cells [21], as well as in bone marrow cells [22]. Shortly after myocardial injury, an increase in AngII concentration occurs, which induces an accumulation, differentiation, and the exit of hematopoietic stem/precursor cells (HPSC) from the bone marrow to contribute to splenic myelopoiesis, supplying up to 50% of the leukocytes to the infarcted area [23], and through the phosphorylation of nuclear factor-kappa B (NF-kB), the binding of AngII to its AT1R receptor induces a pro-inflammatory response mediated by tumor necrosis factor-alpha (TNF-α) or interleukin-1 beta (IL1ß), which in turn are drivers of inflammation [24]. Nevertheless, when neurohumoral response becomes chronic, it leads to an excessive loss of cardiomyocytes, an exacerbated inflammatory response, and the healing and adverse remodeling of the infarcted ventricle, which ultimately underlies HF [25][26][27]. This dysfunctional environment has been proposed to trigger the secretion of several factors into the circulation that can be synthesized in various cell types surrounding the heart and its own cell components, including cardiomyocytes, fibroblasts, smooth muscle (aortic or blood-derived progenitors), and vascular endothelial cells (Figure 1) [28][29].
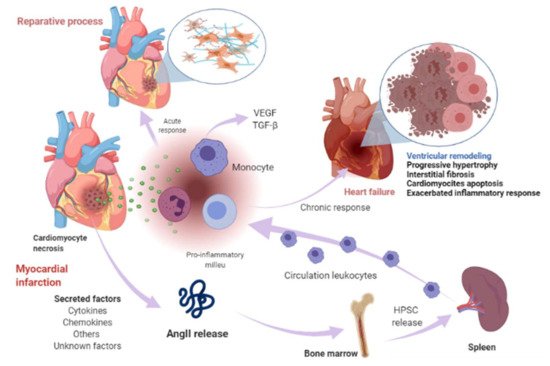
Figure 1. Myocardial infarction and heart-failure-related events. Shortly after myocardial injury, an increase in AngII concentration occurs and induces an accumulation, differentiation, and exit of hematopoietic stem/precursor cells (HPSC) from the bone marrow to contribute to splenic myelopoiesis to supply the infarcted area of the immune cells. Cardiomyocyte necrosis releases signals of danger and induces the secretion of cytokines, chemokines, and adhesion molecules to allow the recruitment and infiltration of leukocytes (mainly monocytes) into the infarcted area. Monocytes exert a reparative response, phagocytosing the cellular debris, while it stimulates repair pathways by secreting pro-inflammatory cytokines through the binding of angiotensin-II (AngII) to type 1 angiotensin-II receptor (AT1R), which induces the phosphorylation of nuclear factor-kappa B (NF-kB). This induces a pro-inflammatory response mediated by tumor necrosis factor-alpha (TNF-α) or interleukin-1 beta (IL1ß) and drives inflammation. The modulation of inflammation in this repair phase includes fibroblast activation and healing mediated in part by renin-angiotensin- aldosterone system (RAAS). When this response becomes chronic, it leads to a pathological process called ventricular remodeling, characterized by progressive hypertrophy of myocytes and interstitial fibrosis, which in later stages involve progressive loss of myocytes through apoptosis, an exacerbated inflammatory response. The healing and the adverse remodeling of the infarcted ventricle ultimately underlie heart failure. This environment can lead to the secretion of certain factors into the circulation that are synthesized in various cell types in the heart, including cardiomyocytes, fibroblasts, smooth muscle, and vascular endothelial cells and other unknown factors. Image created with BioRender.com (Toronto, ON, Canada).
In this scenario, one of these secreted or leading factors can be components of RAAS, especially, AngII. This statement is based on several facts. On one hand, the activation of AngII/AT1R axis is generally associated with the pathophysiological appearance in HF, and it has also been established that RAAS is frequently altered in a variety of cancer types, which in turn is associated with a poor prognosis [30]. It should be considered that the pathological effects observed in these diseases are mainly associated with the AngII/ATR1 axis [12]. Thus, AT1R signaling increases aldosterone levels and blood pressure, induces vasoconstriction, cardiac hypertrophy, fibrosis, inflammation, and reactive oxygen species (ROS) production, while decreasing nitric oxide (NO) production, among other effects [1][9]. In the cancer scenario, AT1R activation by AngII favors cell proliferation, inhibits apoptosis, and promotes adhesion molecule expression, the interaction of monocytes with endothelial cells (EC), the infiltration of inflammatory cells, and the generation of pro-inflammatory cytokines, enabling the establishment of the inflammatory microenvironment, which is a pivotal state for the subsistence of neoplastic cells [14].
On another topic, it has previously been proposed that the initial immune response against a neoplasm is the result of the presence of an acute tissue injury that has generated a chronic infiltration of various myeloid cells, triggering a state of chronic inflammation in the tissue environment because the initial acute inflammatory response did not resolve [31]. Along the same lines, RAAS is a driver of tumorigenesis, linking with HF through immune and inflammatory responses. The involvement of immune cells both in the acute response after MI, the progression towards HF, and in the tumor microenvironment (TME) is a well-established notion. In the TME, immune cells intervene in various stages, mainly due to their infiltration into tumors and their differentiation into tumor-associated macrophages (TAM) [32], which are important components of the infiltration of most tumors and are derived mainly from circulating monocytes and which are attracted to the tumor by chemokines. In such a tumorigenic microenvironment, TAMs can stimulate tumor cell proliferation, promote angiogenesis, and favor invasion and metastasis [33]. In TME, they are frequently located surrounding blood vessels, where they secrete vascular endothelial growth factor (VEGF) and induce new blood vessel formation (angiogenesis). TAMs are the major immunoregulatory cells, and result in immune suppression in TME [34]. It should be noted that in the post-MI phase, monocytes predominate [35][26]. In MI, monocytes secrete angiogenic mediators, such as VEGF and the fibrogenic mediator transforming growth factor-beta (TGF-β), where their function is to promote the repair of infarcted tissue and cardiac fibrosis [26], so these HF-immune mediators can act as a substrate for the development of cancer cells by modulating a favorable microenvironment for its development (Figure 2).
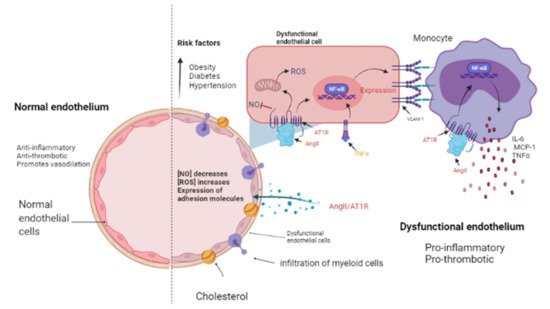
Figure 2. The endothelium in a normal and pathological state. The endothelium is a monolayer of cells that covers the interior of each major and minor vessel. A healthy endothelium has anti-inflammatory, anti-thrombotic properties and promotes vasodilation through nitric oxide (NO) release (left side). Cardiovascular risk factors such as obesity, diabetes and hypertension could promote a dysfunctional endothelium (right side) that is characterized by a decrease in NO release as well as an increment in reactive oxygen species (ROS) and a pro-inflammatory activity mediated by AngII/AT1R signaling, which activates NF-κB and, consequently, the expression of cytokines, chemokines, and adhesion molecules: interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and vascular cell adhesion molecule-1 (VCAM-1) by endothelial cells. Then, myeloid cells such as monocytes migrate and infiltrate towards the aortic walls (where they become macrophages) contributing to the endothelial dysfunction by producing tumor necrosis factor-alpha (TNF-α), IL-6, and MCP-1. Image created with BioRender.com, Toronto, ON, Canada.
Additionally, it was reported that systemic changes induced by the tumor influence the phenotype of circulating monocytes (such as the acquisition of immunosuppressive activity and a decreased responsiveness to inflammatory stimuli) before their infiltration into the tumor environment [32]. In particular, the inflammatory Ly6Chigh monocyte subset is efficiently recruited towards tumors and provides mediators that stimulate cancer-associated inflammation and angiogenesis [23][32]. Consistent with the above, Koelwyn et al. reported that MI accelerates breast cancer growth and cancer-specific mortality in mice and humans. In a murine model, there was an increase in the levels of circulating Ly6Chigh monocytes that were epigenetically reprogrammed in the spleen towards an immunosuppressive phenotype that was maintained in the tumor, as well as in the blood circulation, and additionally demonstrated that the depletion of these cells abolished MI-induced tumor growth [35]. AngII plays a relevant role in macrophage-mediated chronic inflammation by modulating this macrophage amplification program, since according to Retamozo et al. the overproduction of AngII increased macrophage progenitors in the spleen, allowing the extramedullary tissue to supply new macrophages associated with tumors throughout cancer progression in a tumor-bearing animal model. In contrast, blocking AngII production prevented the amplification of macrophage progenitors [23]. In this context, RAAS, and in particular AngII, could be a key point in the convergence of HF and cancer pathophysiology.
That is why it has been stated that chronic inflammation is a point of convergence between HF and cancer; because the former is characterized by chronic inflammation, it directly influences the risk of cancer development in patients with HF, since inflammation is an established component of carcinogenesis. Evidence suggests that chronic inflammation is responsible for up to 25% of all cancers [36]. On this occasion, we want to highlight the role of endothelial dysfunction as a pathophysiological substrate in the development of MI and HF, which in turn can generate an environment conducive to cancer progression.
The presence of cardiovascular risk factors, among them obesity, diabetes, and hypertension, directly affects the endothelium which is composed by EC. These cells line the inside of all major and minor vessels and serve as the first point of contact between the lumen and other tissues and regulate vascular tone, stiffness, inflammation, thrombotic potential in both health and illness (Figure 2). Cardiovascular risk factors mediate their detrimental effects on the vessel wall in part via enhanced activity of RAAS and increased release of vasoactive agents including Ang II as well as paracrine and circulating factors that regulate the generation and activity of endothelium-derived vasoactive and growth factors, adhesion molecules that mediate leucocyte-EC interaction, and blood coagulation regulators [37][38]. The endothelium in a healthy vasculature is anti-inflammatory, anti-thrombotic, and promotes vasodilation but, on the contrary, when the endothelium becomes dysfunctional, it is characterized by a pro-inflammatory and pro-thrombotic state [39][40] (Figure 2). It is worth noting that the exact mechanism by which a normally functioning endothelium becomes dysfunctional remain unknown. However, it has been reported that the endothelium is a prime site for the effects of cardiovascular risk factors; thus, endothelial function can be seen as an integrated index and sensitive measure of cardiovascular disease risk, since it reflects the cumulative contribution of various risk variables associated with inflammation and oxidative stress and given the similar pathological mechanisms that underpin cancer and cardiovascular disease [41]; thus, this is an elemental cellular component that can intermediate the transition between HF and cancer (Figure 2).
From a cancer perspective, dysfunctional ECs can promote pro-inflammatory signaling that is associated with characteristics that favor cancer progression, while in non-pathological conditions, it has been reported that ECs mitigate tumor invasiveness and metastasis [42]. Using in vitro models of dysfunctionally activated ECs, Franses et al. observed that resting EC constructs exhibited moderate inflammatory activity and could inhibit the proliferation and invasion of cancer cells. In contrast, “dysfunctional” ECs favored spontaneous metastasis in adjacent tumors through an aberrant expression of pro-inflammatory cytokines, extracellular matrix, alterations in the leukocyte adhesion process, increasing the expression of vascular cell adhesion molecule-1 (VCAM-1) and abnormal responses to oxidative stress, which are pathological stimuli present both in atherosclerotic lesions, precursors of MI and HF, as well as in the tumor environment (Figure 3) [38][42]. Molitor et al. provided evidence that the AngII/AT1R axis favors the migration and infiltration of myeloid cells towards the aortic walls, inducing endothelial dysfunction. Notably, AT1R blockade with telmisartan attenuated vascular infiltration of immune cells, reducing oxidative stress, and improved endothelial dysfunction [43]. Furthermore, in a subsequent study, they tested ACE inhibition in an HF model after MI, where they observed a decrease in systemic inflammation accompanied by a reduction in vascular infiltration of inflammatory myeloid cells and a decrease in the ROS levels nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) produced [24].
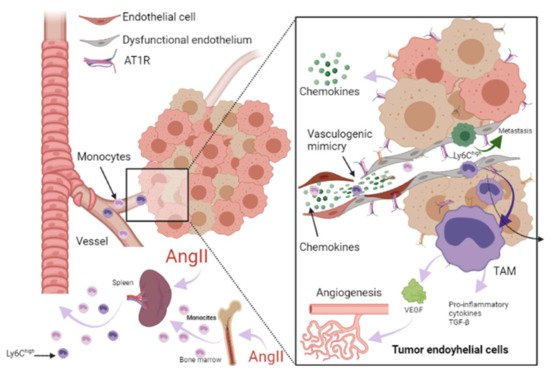
Figure 3. Role of RAAS in the tumor microenvironment. Immune cells can infiltrate tumors and differentiate into tumor-associated macrophages (TAM) derived mainly from circulating monocytes and are attracted to the tumor by chemokines. TAMs can stimulate tumor cell proliferation, angiogenesis, invasion and metastasis. Additionally, tumor microenvironment (TME) can influence the phenotype of circulating monocytes such as the Ly6Chigh monocyte subset, giving them an immunosuppressive activity and a decreased responsiveness to inflammatory stimuli before their infiltration intoTME. Angiotensin-II (AngII) plays a relevant role in macrophage-mediated chronic inflammation, increasing macrophage progenitors and supplying of TAMs. Additionally, endothelial cells (EC) can promote pro-inflammatory signaling, favoring spontaneous metastasis in adjacent tumors through an aberrant expression of pro-inflammatory cytokines, extracellular matrix, alterations in the leukocyte adhesion process, increasing vascular cell adhesion molecule-1 (VCAM-1) and abnormal responses to oxidative stress. In ECs, AngII/AT1R signaling generates a pro-angiogenic response mediated by vascular endothelial growth factor (VEGF). AngII signaling activates TNF-α and NF-κB and upregulates pro-inflammatory endothelial chemokines. AngII has been reported to be able to promote VCAM-1 expression and enhance adhesion, growth, angiogenesis, and the inflammatory microenvironment through AT1R in hepatocellular carcinoma. It has been reported that angiogenesis promotes tumor cell metastasis. Image created with BioRender.com, Toronto, ON, Canada.
ECs are essential in the tumor microenvironment, as they can also express and use components of the RAAS signaling pathway to promote tumor growth, enhance angiogenesis, and promote metastasis [30][44]. It should be noted that adhesion molecules play a crucial role in these processes, since they allow the union and transendothelial migration, in this case, of tumor cells or TAMs. AngII signaling activates TNF-α and NF-κB and upregulates pro-inflammatory endothelial chemokines [38][45]. AngII has been reported to be able to promote VCAM-1 expression and enhance adhesion, growth, angiogenesis, and the inflammatory microenvironment through AT1R in hepatocellular carcinoma [46]. During HF, an increase in VCAM-1 expression was observed in response to AngII stimulation, and this is associated with endothelial dysfunction as well [43][47].
Angiogenesis is involved in myocardial healing. It has been reported to promote tumor progression, depending first on supplying oxygen and nutrients and later, generating a pathway for its metastasis [48]. In ECs, AT1R-mediated AngII generates a pro-angiogenic response mediated by VEGF, a crucial stimulator of pathological vessel formation (Figure 3) [49]. Therefore, this represents another mechanism in which HF could influence the environment that leads to cancer progression. Thus, the study of endothelial dysfunction mechanisms is crucial to preventing the recurrence of serious secondary events in patients who have suffered HF, including the development of cancer.
2. Papel de la farmacoterapia con inhibidores del RAAS en la insuficiencia cardíaca y el cáncer
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) are part of the basic pharmacological treatment after MI (besides reperfusion), and its administration has been reported to reduce mortality in the short and long term in addition to reducing the risk of HF [50][51][52]. Additionally, recent data suggest that these drugs also possess anti-inflammatory and anticancer characteristics [53][54], which reinforces the convergent role of the RAAS system in HF and cancer, because their molecular inhibitors play an important role in development, migration, recurrence, and resistance to antineoplastic drugs [54].
To denote the role that RAAS plays in HF and cancer, we want to highlight the therapeutic properties that RAAS inhibitors possess in HF (Table 1) and the findings that these inhibitors have shown in cancer preclinical research (Table 2).
Table 1. Role of RAAS inhibitors in heart failure.
| RAAS Inhibitor | Observations |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Long-term administration was associated with an improvement in survival and reduced morbidity and mortality due to major cardiovascular events in patients with asymptomatic left ventricular (LV) dysfunction after myocardial infarction (MI) [55]. |
| Enalapril | Increased exercise time and left ventricular ejection fraction (LVEF) [56]. |
| Perindopril | Increased 6 min walk distance but did not decrease mortality [57]. After 1-year treatment reduced progressive LV remodeling but it was not associated with better clinical outcomes [58]. |
| Ramipril | Administration to patients with clinical evidence of either transient or ongoing heart failure (HF) after MI resulted in a substantial reduction in premature death from all causes [59]. |
| Trandolapril | Long-term treatment in patients with reduced LV function soon after MI significantly reduced the risk of overall mortality, mortality from cardiovascular causes, sudden death, and the development of severe HF [60]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Telmisartan was well tolerated in patients unable to tolerate ACEI. Although the drug had no significant effect on hospitalizations for HF, it modestly reduced the risk of the composite outcome of cardiovascular death, MI, or stroke [61]. |
| Candesartan | Slightly decreased hospitalizations but did not decrease mortality [62]. Reduced cardiovascular mortality and hospital admissions for worsening chronic HF. Patients with reduced ejection fraction were the most benefited [63]. |
| Losartan | Reduced the rate of death or admission for HF in patients with HF, reduced LVEF, and intolerance to ACEI [64]. |
| Valsartan | In patients with MI associated with HF and/or LV dysfunction, valsartan administration in the immediate post MI period demonstrated equal efficacy than captopril [65][66]. |
| Aldosterone antagonists | |
| Spironolactone | Prevented LV fibrosis and remodeling after MI [67] |
Table 2. Role of RAAs inhibitor in cancer preclinical research.
| RAAS Inhibitor | Findings |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Inhibits tumor growth in a gastric cancer model and suppresses the angiogenesis of the tumor by decreasing the expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP)-7 in a mouse model with human gastric cancer [68]. Attenuates cell migration in a breast cancer model [69]. Inhibits cell growth, decreases c-myc expression, and increases apoptosis on leukemic cell lines [70]. |
| Enalapril | Inhibits tumor progression and reduces number of tumor-associated macrophages (TAMs) [23]. |
| Perindopril | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor by decreasing the expression of VEGF and MMP-7 in a mouse model with human gastric cancer [68]. |
| Ramipril | Decreases systemic inflammation [24]. |
| Trandolapril | Inhibits cell growth, decreases c-myc expression, and increases apoptosis in leukemic cell lines [70]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Inhibits cell proliferation and tumor growth of esophageal squamous cell carcinoma by inducing s-phase cell cycle arrest [71]. |
| Candesartan | Prevents bladder cancer growth in a mouse model by inhibiting angiogenesis, and combined treatment with candesartan and paclitaxel enhances paclitaxel-induced cytotoxicity [72]. Candesartan treatment significantly sensitizes human lung adenocarcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis [73]. |
| Losartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor decreasing the expressions of VEGF [68]. Can exert anti-metastatic activity by inhibiting chemokine receptor type 2 (CCR2) signaling and suppressing monocyte recruitment in a mouse model with tumors and indirectly as anti-inflammatory effect and independently of AT1R [74]. Ameliorates angiogenesis, inflammation and the induction of oxidative stress via type-1 angiotensin-II receptor (AT1R) in a murine model of lung metastasis of colorectal cancer [75]. Inhibits cell growth, decreases c-myc expression and increases apoptosis in leukemic cell lines [70]. |
| Valsartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor, decreasing the expressions of VEGF [68]. |
| Aldosterone antagonists | |
| Spironolactone | Inhibits cancerous cell growth and is highly toxic for cancer stem cells; impairs DNA-double-strand breaks repair and induces apoptosis in cancer cells and cancer stem cells (CSCs) while sparing healthy cells. In vivo, this treatment reduces the size and CSC content of tumors [76]. |
References
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129.
- Pugliese, N.R.; Masi, S.; Taddei, S. The renin-angiotensin-aldosterone system: A crossroad from arterial hypertension to heart failure. Heart Fail. Rev. 2020, 25, 31–42.
- Ziaja, M.; Urbanek, K.A.; Kowalska, K.; Piastowska-Ciesielska, A.W. Angiotensin II and Angiotensin Receptors 1 and 2—Multifunctional System in Cells Biology, What Do We Know? Cells 2021, 10, 381.
- Steckelings, U.M.; Paulis, L.; Namsolleck, P.; Unger, T. AT2 receptor agonists: Hypertension and beyond. Curr. Opin. Nephrol. Hypertens. 2012, 21, 142–146.
- Wang, Y.; Del Borgo, M.; Lee, H.W.; Baraldi, D.; Hirmiz, B.; Gaspari, T.A.; Denton, K.M.; Aguilar, M.I.; Samuel, C.S.; Widdop, R.E. Anti-fibrotic potential of AT2 receptor agonists. Front. Pharmacol. 2017, 8, 564.
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor κb. Hypertension 2010, 55, 924–931.
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, eaan5616.
- Sheikh, S.P. Angiotensin Type 2 Receptor. In Encyclopedia of Signaling Molecules; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 320–327.
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin system in heart failure. Circ. Res. 2016, 118, 1313–1326.
- Wang, J.; He, W.; Guo, L.; Zhang, Y.; Li, H.; Han, S.; Shen, D. The ACE2-Ang (1-7)-Mas receptor axis attenuates cardiac remodeling and fibrosis in post-myocardial infarction. Mol. Med. Rep. 2017, 16, 1973–1981.
- da Silveira, K.D.; Coelho, F.M.; Vieira, A.T.; Sachs, D.; Barroso, L.C.; Costa, V.V.; Bretas, T.L.B.; Bader, M.; de Sousa, L.P.; da Silva, T.A.; et al. Anti-Inflammatory Effects of the Activation of the Angiotensin-(1–7) Receptor, Mas, in Experimental Models of Arthritis. J. Immunol. 2010, 185, 5569–5576.
- Chappell, M.C.; Al Zayadneh, E.M. Angiotensin-(1-7) and the Regulation of Anti-Fibrotic Signaling Pathways. J. Cell Signal. 2017, 2, 2.
- Park, B.M.; Cha, S.A.; Lee, S.H.; Kim, S.H. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides 2016, 79, 66–74.
- de Paula Gonzaga, A.L.A.C.; Palmeira, V.A.; Ribeiro, T.F.S.; Costa, L.B.; de Sá Rodrigues, K.E.; Simões-e-Silva, A.C. ACE2/Angiotensin-(1-7)/Mas Receptor Axis in Human Cancer: Potential Role for Pediatric Tumors. Curr. Drug Targets 2020, 21, 892–901.
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738.
- Bertero, E.; Ameri, P.; Maack, C. Bidirectional Relationship Between Cancer and Heart Failure: Old and New Issues in Cardio-oncology. Card. Fail. Rev. 2019, 5, 106–111.
- Emdin, M.; Fatini, C.; Mirizzi, G.; Poletti, R.; Borrelli, C.; Prontera, C.; Latini, R.; Passino, C.; Clerico, A.; Vergaro, G. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: How useful, how feasible? Clin. Chim. Acta 2015, 443, 85–93.
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329.
- Kyaw, T.; Loveland, P.; Kanellakis, P.; Cao, A.; Kallies, A.; Huang, A.L.; Peter, K.; Toh, B.-H.; Bobik, A. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 2021, 42, 938–947.
- Orsborne, C.; Chaggar, P.S.; Shaw, S.M.; Williams, S.G. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: The past, the present and the future. Postgrad. Med. J. 2016, 93, 29–37.
- Rasini, E.; Cosentino, M.; Marino, F.; Legnaro, M.; Ferrari, M.; Guasti, L.; Venco, A.; Lecchini, S. Angiotensin II type 1 receptor expression on human leukocyte subsets: A flow cytometric and RT-PCR study. Regul. Pept. 2006, 134, 69–74.
- Kim, S.; Zingler, M.; Harrison, J.K.; Scott, E.W.; Cogle, C.R.; Luo, D.; Raizada, M.K. Angiotensin II Regulation of Proliferation, Differentiation, and Engraftment of Hematopoietic Stem Cells. Hypertension 2016, 67, 574–584.
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Ryan, R.; Pucci, F.; Sio, S.W.; Kuswanto, W.; Rauch, P.J.; Chudnovskiy, A.; Iwamoto, Y.; et al. Angiotensin II Drives the Production of Tumor-Promoting Macrophages. Immunity 2013, 38, 296–308.
- Rudi, W.-S.; Molitor, M.; Garlapati, V.; Finger, S.; Wild, J.; Münzel, T.; Karbach, S.H.; Wenzel, P. ACE Inhibition Modulates Myeloid Hematopoiesis after Acute Myocardial Infarction and Reduces Cardiac and Vascular Inflammation in Ischemic Heart Failure. Antioxidants 2021, 10, 396.
- Frangogiannis, N.G. The immune system and the remodeling infarcted heart: Cell biological insights and therapeutic opportunities. J. Cardiovasc. Pharmacol. 2014, 63, 185–195.
- Libby, P.; Nahrendorf, M.; Swirski, F.K. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease an expanded cardiovascular continuum. J. Am. Coll. Cardiol. 2016, 67, 1091–1103.
- Katz, S.D. Mechanisms of Heart Failure BT—Management of Heart Failure: Volume 1: Medical; Baliga, R.R., Haas, G.J., Eds.; Springer: London, UK, 2015; pp. 13–30. ISBN 978-1-4471-6657-3.
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; De Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; Van Der Vegt, B.; et al. Heart failure stimulates tumor growth by circulating factors. Circulation 2018, 138, 678–691.
- Dewey, C.M.; Spitler, K.M.; Ponce, J.M.; Hall, D.D.; Grueter, C.E. Cardiac-Secreted Factors as Peripheral Metabolic Regulators and Potential Disease Biomarkers. J. Am. Heart Assoc. 2016, 5, e003101.
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin-angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759.
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527.
- Kiss, M.; Caro, A.A.; Raes, G.; Laoui, D. Systemic Reprogramming of Monocytes in Cancer. Front. Oncol. 2020, 10, 1399.
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126.
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925.
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458.
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752.
- Becher, U.M.; Endtmann, C.; Tiyerili, V.; Nickenig, G.; Werner, N. Endothelial damage and regeneration: The role of the renin-angiotensin- aldosterone system. Curr. Hypertens. Rep. 2011, 13, 86–92.
- Silva, G.M.; França-Falcão, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1–7. Front. Physiol. 2020, 11, 1067.
- Davel, A.P.; Anwar, I.J.; Jaffe, I.Z. The endothelial mineralocorticoid receptor: Mediator of the switch from vascular health to disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 97–104.
- Herrera-Zelada, N.; Zuñiga-Cuevas, U.; Ramirez-Reyes, A.; Lavandero, S.; Riquelme, J.A. Targeting the Endothelium to Achieve Cardioprotection. Front. Pharmacol. 2021, 12, 3.
- Toya, T.; Sara, J.D.; Corban, M.T.; Taher, R.; Godo, S.; Herrmann, J.; Lerman, L.O.; Lerman, A. Assessment of peripheral endothelial function predicts future risk of solid-tumor cancer. Eur. J. Prev. Cardiol. 2020, 27, 608–618.
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344.
- Molitor, M.; Rudi, W.S.; Garlapati, V.; Finger, S.; Schüler, R.; Kossmann, S.; Lagrange, J.; Nguyen, T.S.; Wild, J.; Knopp, T.; et al. Nox2+myeloid cells drive vascular inflammation and endothelial dysfunction in heart failure after myocardial infarction via angiotensin II receptor type 1. Cardiovasc. Res. 2021, 117, 162–177.
- Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Cordeiro, C.R.; Medeiros, R. Renin-angiotensin system in lung tumor and microenvironment interactions. Cancers 2020, 12, 1457.
- Ishikane, S.; Takahashi-Yanaga, F. The role of angiotensin II in cancer metastasis: Potential of renin-angiotensin system blockade as a treatment for cancer metastasis. Biochem. Pharmacol. 2018, 151, 96–103.
- Feng, L.-H.; Sun, H.-C.; Zhu, X.-D.; Zhang, S.-Z.; Li, X.-L.; Li, K.-S.; Liu, X.-F.; Lei, M.; Li, Y.; Tang, Z.-Y. Irbesartan inhibits metastasis by interrupting the adherence of tumor cell to endothelial cell induced by angiotensin II in hepatocellular carcinoma. Ann. Transl. Med. 2021, 9, 207.
- Patel, R.B.; Colangelo, L.A.; Bielinski, S.J.; Larson, N.B.; Ding, J.; Allen, N.B.; Michos, E.D.; Shah, S.J.; Lloyd-Jones, D.M. Circulating Vascular Cell Adhesion Molecule-1 and Incident Heart Failure: The Multi-Ethnic Study of Atherosclerosis (MESA). J. Am. Heart Assoc. 2020, 9, e019390.
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millán, S.; Sánchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. JRAAS J. Renin Angiotensin Aldosterone Syst. 2015, 16, 227–233.
- Carbajo-Lozoya, J.; Lutz, S.; Feng, Y.; Kroll, J.; Hammes, H.P.; Wieland, T. Angiotensin II modulates VEGF-driven angiogenesis by opposing effects of type 1 and type 2 receptor stimulation in the microvascular endothelium. Cell. Signal. 2012, 24, 1261–1269.
- Cespón-Fernández, M.; Raposeiras-Roubín, S.; Abu-Assi, E.; Manzano-Fernández, S.; Flores-Blanco, P.; Barreiro-Pardal, C.; Castiñeira-Busto, M.; Muñoz-Pousa, I.; López-Rodríguez, E.; Caneiro-Queija, B.; et al. Renin–Angiotensin System Blockade and Risk of Heart Failure After Myocardial Infarction Based on Left Ventricular Ejection Fraction: A Retrospective Cohort Study. Am. J. Cardiovasc. Drugs 2019, 19, 487–495.
- Raposeiras-Roubín, S.; Abu-Assi, E.; Cespón-Fernández, M.; Ibáñez, B.; García-Ruiz, J.M.; D’Ascenzo, F.; Simao Henriques, J.P.; Saucedo, J.; Caneiro-Queija, B.; Cobas-Paz, R.; et al. Impact of renin-angiotensin system blockade on the prognosis of acute coronary syndrome based on left ventricular ejection fraction. Rev. Esp. Cardiol. 2020, 73, 114–122.
- Sim, H.W.; Zheng, H.; Richards, A.M.; Chen, R.W.; Sahlen, A.; Yeo, K.K.; Tan, J.W.; Chua, T.; Tan, H.C.; Yeo, T.C.; et al. Beta-blockers and renin-angiotensin system inhibitors in acute myocardial infarction managed with inhospital coronary revascularization. Sci. Rep. 2020, 10, 15184.
- Ranjbar, R.; Shafiee, M.; Hesari, A.R.; Ferns, G.A.; Ghasemi, F.; Avan, A. The potential therapeutic use of renin–angiotensin system inhibitors in the treatment of inflammatory diseases. J. Cell. Physiol. 2019, 234, 2277–2295.
- Yang, R.; Zhang, Y.; Liao, X.; Yao, Y.; Huang, C.; Liu, L. The Relationship Between Anti-Hypertensive Drugs and Cancer: Anxiety to be Resolved in Urgent. Front. Pharmacol. 2020, 11, 2122.
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of Captopril on Mortality and Morbidity in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 1992, 327, 669–677.
- Aronow, W.S.; Kronzon, I. Effect of enalapril on congestive heart failure treated with diuretics in elderly patients with prior myocardial infarction and normal left ventricular ejection fraction. Am. J. Cardiol. 1993, 71, 602–604.
- Cleland, J.G.F.; Tendera, M.; Adamus, J.; Freemantle, N.; Gray, C.S.; Lye, M.; O’Mahony, D.; Polonski, L.; Taylor, J. Perindopril for elderly people with chronic heart failure: The PEP-CHF study. Eur. J. Heart Fail. 1999, 1, 211–217.
- Ferrari, R. Effects of angiotensin-converting enzyme inhibition with perindopril on left ventricular remodeling and clinical outcome: Results of the randomized Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) study. Arch. Intern. Med. 2006, 166, 659–666.
- The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet 1993, 342, 821–828.
- Tepper, D.; Greenberg, S. A clinical trial of the angiotensin-converting enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Cardiovasc. Rev. Rep. 1996, 17, 49.
- The Telmisartan Randomised AssessmeNt Study in ACE iNtolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators; Yusuf, S.; Teo, K.; Anderson, C.; Pogue, J.; Dyal, L.; Copland, I.; Schumacher, H.; Dagenais, G.; Sleight, P. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet 2008, 372, 1174–1183.
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.V.; Michelson, E.L.; Olofsson, B.; Östergren, J. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-preserved trial. Lancet 2003, 362, 777–781.
- Pfeffer, M.A.; McMurray, J.J.; Östergren, J.; Granger, C.B.; Yusuf, S.; Pitt, B. Candesartan reduced mortality and hospital admissions in chronic heart failure. Evid. Based. Med. 2004, 9, 44–45.
- Konstam, M.A.; Neaton, J.D.; Dickstein, K.; Drexler, H.; Komajda, M.; Martinez, F.A.; Riegger, G.A.; Malbecq, W.; Smith, R.D.; Guptha, S.; et al. Effects of high-dose versus low-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): A randomised, double-blind trial. Lancet 2009, 374, 1840–1848.
- Pfeffer, M.A.; McMurray, J.; Leizorovicz, A.; Maggioni, A.P.; Rouleau, J.L.; Van De Werf, F.; Henis, M.; Neuhart, E.; Gallo, P.; Edwards, S.; et al. Valsartan in acute myocardial infarction trial (VALIANT): Rationale and design. Am. Heart J. 2000, 140, 727–750.
- Bissessor, N.; White, H. Valsartan in the treatment of heart failure or left ventricular dysfunction after myocardial infarction. Vasc. Health Risk Manag. 2007, 3, 425–430.
- Hayashi, M.; Tsutamoto, T.; Wada, A.; Tsutsui, T.; Ishii, C.; Ohno, K.; Fujii, M.; Taniguchi, A.; Hamatani, T.; Nozato, Y.; et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003, 107, 2559–2565.
- Wang, L.; Cai, S.R.; Zhang, C.H.; He, Y.L.; Zhan, W.H.; Wu, H.; Peng, J.J. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor blockers on lymphangiogenesis of gastric cancer in a nude mouse model. Chin. Med. J. 2008, 121, 2167–2171.
- Rasha, F.; Kahathuduwa, C.; Ramalingam, L.; Hernandez, A.; Moussa, H.; Moustaid-Moussa, N. Combined Effects of Eicosapentaenoic Acid and Adipocyte Renin–Angiotensin System Inhibition on Breast Cancer Cell Inflammation and Migration. Cancers 2020, 12, 220.
- De la Iglesia Iñigo, S.; López-Jorge, C.E.; Gómez-Casares, M.T.; Lemes Castellano, A.; Martín Cabrera, P.; López Brito, J.; Suárez Cabrera, A.; Molero Labarta, T. Induction of apoptosis in leukemic cell lines treated with captopril, trandolapril and losartan: A new role in the treatment of leukaemia for these agents. Leuk. Res. 2009, 33, 810–816.
- Matsui, T.; Chiyo, T.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Nakahara, M.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Telmisartan Inhibits Cell Proliferation and Tumor Growth of Esophageal Squamous Cell Carcinoma by Inducing S-Phase Arrest In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3197.
- Kosugi, M.; Miyajima, A.; Kikuchi, E.; Kosaka, T.; Horiguchi, Y.; Murai, M. Effect of angiotensin II type 1 receptor antagonist on tumor growth and angiogenesis in a xenograft model of human bladder cancer. Hum. Cell 2007, 20, 1–9.
- Rasheduzzaman, M.; Park, S.Y. Antihypertensive drug-candesartan attenuates TRAIL resistance in human lung cancer via AMPK-mediated inhibition of autophagy flux. Exp. Cell Res. 2018, 368, 126–135.
- Regan, D.P.; Coy, J.W.; Chahal, K.K.; Chow, L.; Kurihara, J.N.; Guth, A.M.; Kufareva, I.; Dow, S.W. The Angiotensin Receptor Blocker Losartan Suppresses Growth of Pulmonary Metastases via AT1R-Independent Inhibition of CCR2 Signaling and Monocyte Recruitment. J. Immunol. 2019, 202, 3087–3102.
- Hashemzehi, M.; Naghibzadeh, N.; Asgharzadeh, F.; Mostafapour, A.; Hassanian, S.M.; Ferns, G.A.; Cho, W.C.; Avan, A.; Khazaei, M. The therapeutic potential of losartan in lung metastasis of colorectal cancer. EXCLI J. 2020, 19, 927–935.
- Gold, A.; Eini, L.; Nissim-Rafinia, M.; Viner, R.; Ezer, S.; Erez, K.; Aqaqe, N.; Hanania, R.; Milyavsky, M.; Meshorer, E.; et al. Spironolactone inhibits the growth of cancer stem cells by impairing DNA damage response. Oncogene 2019, 38, 3103–3118.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
5 times
(View History)
Update Date:
16 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No