1. Sistema renina-angiotensina-aldosterona que vincula la insuficiencia cardíaca isquémica y el cáncerRenin-Angiotensin-Aldosterone System Linking Ischemic Heart Failure and Cancer
Hay dos vías principales en el RAAS: vías clásicas y no clásicas. En el RAAS clásico, el péptido efector es la angiotensina-II (AngII), que se produce a partir de su precursor hepático, el angiotensinógeno, que es catabolizado por la enzima renina, dando a su vez a la angiotensina-I (AngI ), que es un sustrato. para la enzima convertidora de angiotensina (ECA) que produce angiotensina-II. Los efectos funcionales de AngII en el RAAS clásicos están mediados en gran medida por el receptor de angiotensina II de tipo 1 (AT1R) y el receptor de tipo 2 (AT2R) [ 12 , 35 ]. La activación de AT1R aumenta la aldosterona, un actor importante en la regulación del equilibrio electrolítico [ 36], pero también la activación de AT1R tiene muchos otros efectos (descritos más adelante). La señalización mediada por el AT2R se asocia con funciones antifibróticas e incluso con efectos antiinflamatorios en la IC [ 37 , 38 , 39 ], mientras que en el cáncer este eje tiene efectos antiproliferativos, antiangiogénicos y proapoptóticos [ 40 , 41 ]. Sin embargo, también hay informes contradictorios que sugirieron posibles diferencias específicas del tipo de tumor [ 36 , 40 ].
There is evidence that the RAAS is involved in most of the tumorigenic characteristics described above, and due to the chronic activation of RAAS in HF, it has been proposed that a failing heart may be closely related to the development of cancer. In this review, we will discuss recent studies that highlight the role of RAAS components as an axis of crucial importance in the pathophysiology of HF and as well as evidence of the dysregulation of its components in the development of cancer to highlight the points where these two entities that were previously considered independent could now converge.
There are two major pathways in the RAAS: classical and non-classical pathways. In the classical RAAS, the effector peptide is angiotensin-II (AngII), which is produced from its hepatic precursor, angiotensinogen, which is catabolized by the enzyme renin, giving rise to angiotensin-I (AngI) in turn, which is a substrate for the angiotensin-converting enzyme (ACE) producing angiotensin-II. The functional effects of AngII in the classical RAAS are largely mediated by the type 1 angiotensin-II receptor (AT1R) and the type 2 receptor (AT2R) [12,35]. AT1R activation increases aldosterone, an important player in the regulation of electrolyte balance [36], but AT1R activation also has many other effects (described later). Signaling mediated by the AT2R is associated with antifibrotic functions and even with anti-inflammatory effects in HF [37,38,39], while in cancer, this axis has antiproliferative, antiangiogenic, and pro-apoptotic effects [40,41]. However, there are also conflicting reports suggesting possible tumor type-specific differences [36,40].
In the non-classical RAAS, the homologue of ACE, angiotensin-converting enzyme 2 (ACE2) cleaves AngI into a nonapeptide, Ang 1-9 and AngII into a heptapeptide, Ang 1-7. Additionally, AngII can be also converted to Ang 2-8 (AngIII) by aminopeptidase A, and exerts its effects by binding to AT1R. Aminopeptidase N converts AngIII to Ang 3-8 (AngIV) and can act through the angiotensin 4 receptor (AT4R) [12]. Ang 1-9 can activate AT2R, and Ang 1-7 can bind to the proto-oncogene Mas receptor (MasR). Interestingly, every one of these components has been demonstrated to counteract the actions of the classical RAAS [12,42]. Signaling mediated by the ACE2/Ang 1-7/MasR axis has been shown to have a protective role in the development of myocardial remodeling post-MI in an animal model [43], but it is also associated with antifibrotic and anti-inflammatory effects [44,45]. Moreover, AngIV/AT4R signaling has a cardioprotective role, acting as a counterpart of Ang II-mediated inflammation and myocardial fibrosis in rat model [46]. In cancer, MasR has been documented to reduce abnormal angiogenesis, inflammation and cell proliferation by the local decrease of Ang II levels or AT1 receptor blockade associated with high concentrations of Ang(1-7) at the tumor site [47]. Even so, as the AT1R continues to be crucial in mediating physiological and pathophysiological effects of AngII [48], in this review, we are going to focus in the classical AT1R/AngII RAAS axis.
AngII overproduction is linked to the development of chronic illnesses; in fact, a chronic activation of RAAS is a hallmark of HF, especially marked by a systemic increase in levels of AngII [10,49], and to better understand how RAAS is implied in both of these diseases, first we must consider the involved pathophysiology from MI to HF and then later to cancer.
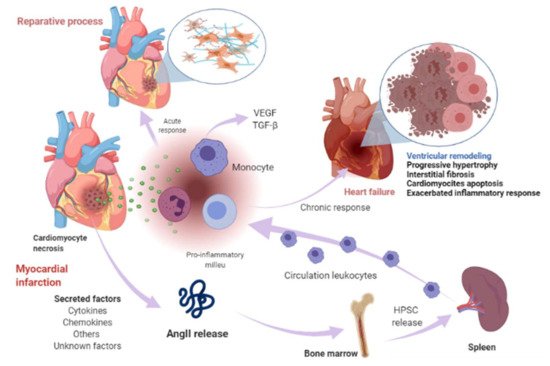
Cardiomyocyte necrosis in the infarcted myocardium activates the innate immune response, triggering an inflammatory response. The release of danger signals from dying cells induces the secretion of cytokines, like chemokines and adhesion molecules, to allow the recruitment and infiltration of leukocytes, mainly monocytes, into the infarcted area, where they exert a “reparative” response, phagocytosing the cellular debris, while stimulating repair pathways by secreting pro-inflammatory cytokines. To supply the appropriate number of immune cells, a release of stem cells and hematopoietic progenitors from the niches of the bone marrow occurs; these cells then migrate to the spleen and, ultimately, increase the production of immune cells, which in turn mediates an effective inflammatory response (Figure 1) [50,51]. The modulation of inflammation in this repair phase includes fibroblast activation and healing mediated by the neurohumoral response. RAAS, which is part of the neurohumoral response, is activated by renal hypoperfusion and sympathetic activation as compensatory mechanisms after a myocardial injury [11]. In these processes, RAAS actively participates mainly through the AngII effector peptide. Indeed, various components of RAAS, including angiotensinogen, AngII, ACE, AT1R, and AT2R, have been reported to be expressed in a variety of immune cells [52], as well as in bone marrow cells [53]. Shortly after myocardial injury, an increase in AngII concentration occurs, which induces an accumulation, differentiation, and the exit of hematopoietic stem/precursor cells (HPSC) from the bone marrow to contribute to splenic myelopoiesis, supplying up to 50% of the leukocytes to the infarcted area [54], and through the phosphorylation of nuclear factor-kappa B (NF-kB), the binding of AngII to its AT1R receptor induces a pro-inflammatory response mediated by tumor necrosis factor-alpha (TNF-α) or interleukin-1 beta (IL1ß), which in turn are drivers of inflammation [55]. Nevertheless, when neurohumoral response becomes chronic, it leads to an excessive loss of cardiomyocytes, an exacerbated inflammatory response, and the healing and adverse remodeling of the infarcted ventricle, which ultimately underlies HF [56,57,58]. This dysfunctional environment has been proposed to trigger the secretion of several factors into the circulation that can be synthesized in various cell types surrounding the heart and its own cell components, including cardiomyocytes, fibroblasts, smooth muscle (aortic or blood-derived progenitors), and vascular endothelial cells (Figure 1) [33,59].
En el RAAS no clásico, el homólogo de ACE, la enzima convertidora de angiotensina 2 (ACE2) escinde AngI en un nonapéptido, Ang 1-9 y AngII en un heptapéptido, Ang 1-7. Además, AngII también se puede convertir en Ang 2-8 (AngIII) por la aminopeptidasa A, y ejerce sus efectos al unirse a AT1R. La aminopeptidasa N convierte AngIII en Ang 3-8 (AngIV) y puede actuar a través del receptor de angiotensina 4 (AT4R) [ 12 ]. Ang 1-9 puede activar AT2R, y Ang 1-7 puede unirse al receptor del protooncogén Mas (MasR). Curiosamente, se ha demostrado que cada uno de estos componentes contrarresta las acciones del RAAS clásico [ 12 , 42]. Se ha demostrado que la señalización mediada por el eje ACE2 / Ang 1-7 / MasR tiene un papel protector en el desarrollo de la remodelación del miocardio después de un IM en un modelo animal [ 43 ], pero también se asocia con efectos antifibróticos y antiinflamatorios [ 44 , 45 ]. Además, la señalización de AngIV / AT4R tiene un papel cardioprotector, actuando como contraparte de la inflamación mediada por Ang II y la fibrosis miocárdica en el modelo de rata [ 46 ]. En el cáncer, se ha documentado que MasR reduce la angiogénesis anormal, la inflamación y la proliferación celular mediante la disminución local de los niveles de Ang II o el bloqueo del receptor AT1 asociado con altas concentraciones de Ang (1-7) en el sitio del tumor [ 47]. Aun así, dado que el AT1R sigue siendo crucial en la mediación de los efectos fisiológicos y fisiopatológicos de la AngII [ 48 ], en esta revisión nos vamos a centrar en el eje clásico AT1R / AngII RAAS.
La sobreproducción de AngII está relacionada con el desarrollo de enfermedades crónicas; de hecho, una activación crónica del RAAS es un sello distintivo de la IC, especialmente marcada por un aumento sistémico en los niveles de AngII [ 10 , 49 ], y para comprender mejor cómo el RAAS está implicado en ambas enfermedades, primero debemos considerar los implicados. fisiopatología de MI a HF y luego al cáncer.
La necrosis de cardiomiocitos en el miocardio infartado activa la respuesta inmune innata, desencadenando una respuesta inflamatoria. La liberación de señales de peligro de las células moribundas induce la secreción de citocinas, como quimiocinas y moléculas de adhesión, para permitir el reclutamiento e infiltración de leucocitos, principalmente monocitos, en el área infartada, donde ejercen una respuesta "reparadora", fagocitando los restos celulares. , mientras estimula las vías de reparación mediante la secreción de citocinas proinflamatorias. Para suministrar la cantidad adecuada de células inmunitarias, se produce una liberación de células madre y progenitores hematopoyéticos de los nichos de la médula ósea; estas células luego migran al bazo y, en última instancia, aumentan la producción de células inmunes, que a su vez media una respuesta inflamatoria eficaz ( Figura 1 ) [50 , 51 ]. La modulación de la inflamación en esta fase de reparación incluye la activación y curación de fibroblastos mediada por la respuesta neurohumoral. El SRAA, que forma parte de la respuesta neurohumoral, se activa por hipoperfusión renal y activación simpática como mecanismos compensatorios después de una lesión miocárdica [ 11 ]. En estos procesos, RAAS participa activamente principalmente a través del péptido efector AngII. De hecho, se ha informado que varios componentes de RAAS, incluidos angiotensinógeno, AngII, ACE, AT1R y AT2R, se expresan en una variedad de células inmunes [ 52 ], así como en células de la médula ósea [ 53]. Poco después de la lesión miocárdica, se produce un aumento en la concentración de AngII, que induce la acumulación, diferenciación y salida de células madre / precursoras hematopoyéticas (HPSC) de la médula ósea para contribuir a la mielopoyesis esplénica, suministrando hasta el 50% de los leucocitos a el área infartada [ 54 ], ya través de la fosforilación del factor nuclear kappa B (NF-kB), la unión de AngII a su receptor AT1R induce una respuesta proinflamatoria mediada por el factor de necrosis tumoral alfa (TNF-α) o interleucina-1 beta (IL1ß), que a su vez son impulsoras de la inflamación [ 55]. Sin embargo, cuando la respuesta neurohumoral se vuelve crónica, conduce a una pérdida excesiva de cardiomiocitos, una respuesta inflamatoria exacerbada y la curación y remodelación adversa del ventrículo infartado, que finalmente subyace a la IC [ 56 , 57 , 58 ]. Se ha propuesto que este entorno disfuncional desencadena la secreción de varios factores en la circulación que pueden sintetizarse en varios tipos de células que rodean el corazón y sus propios componentes celulares, incluidos cardiomiocitos, fibroblastos, músculo liso (progenitores aórticos o derivados de la sangre) y células endoteliales vasculares ( Figura 1 ) [ 33 , 59 ].
Figura 1. Infarto de miocardio y eventos relacionados con insuficiencia cardíaca. Poco después de la lesión miocárdica, se produce un aumento de la concentración de AngII e induce la acumulación, diferenciación y salida de células madre / precursoras hematopoyéticas (HPSC) de la médula ósea para contribuir a la mielopoyesis esplénica para irrigar el área infartada de las células inmunitarias. La necrosis de cardiomiocitos libera señales de peligro e induce la secreción de citocinas, quimiocinas y moléculas de adhesión para permitir el reclutamiento e infiltración de leucocitos (principalmente monocitos) en el área infartada. Los monocitos ejercen una respuesta reparadora, fagocitosan los restos celulares, mientras que estimulan las vías de reparación secretando citocinas proinflamatorias a través de la unión de la angiotensina-II (AngII) al receptor de angiotensina-II tipo 1 (AT1R), que induce la fosforilación del factor nuclear kappa B (NF-kB). Esto induce una respuesta proinflamatoria mediada por el factor de necrosis tumoral alfa (TNF-α) o interleucina-1 beta (IL1ß) e impulsa la inflamación. La modulación de la inflamación en esta fase de reparación incluye la activación y curación de fibroblastos mediada en parte por el sistema renina-angiotensina-aldosterona (RAAS). Cuando esta respuesta se vuelve crónica, conduce a un proceso patológico llamado remodelado ventricular, caracterizado por hipertrofia progresiva de miocitos y fibrosis intersticial, que en etapas posteriores implica pérdida progresiva de miocitos por apoptosis, una respuesta inflamatoria exacerbada. La curación y la remodelación adversa del ventrículo infartado subyacen en última instancia a la insuficiencia cardíaca. Este entorno puede conducir a la secreción de ciertos factores en la circulación que se sintetizan en varios tipos de células en el corazón, incluidos cardiomiocitos, fibroblastos, músculo liso y células endoteliales vasculares y otros factores desconocidos. Imagen creada con BioRender.com (Toronto, ON, Canadá).Figure 1. Myocardial infarction and heart-failure-related events. Shortly after myocardial injury, an increase in AngII concentration occurs and induces an accumulation, differentiation, and exit of hematopoietic stem/precursor cells (HPSC) from the bone marrow to contribute to splenic myelopoiesis to supply the infarcted area of the immune cells. Cardiomyocyte necrosis releases signals of danger and induces the secretion of cytokines, chemokines, and adhesion molecules to allow the recruitment and infiltration of leukocytes (mainly monocytes) into the infarcted area. Monocytes exert a reparative response, phagocytosing the cellular debris, while it stimulates repair pathways by secreting pro-inflammatory cytokines through the binding of angiotensin-II (AngII) to type 1 angiotensin-II receptor (AT1R), which induces the phosphorylation of nuclear factor-kappa B (NF-kB). This induces a pro-inflammatory response mediated by tumor necrosis factor-alpha (TNF-α) or interleukin-1 beta (IL1ß) and drives inflammation. The modulation of inflammation in this repair phase includes fibroblast activation and healing mediated in part by renin-angiotensin- aldosterone system (RAAS). When this response becomes chronic, it leads to a pathological process called ventricular remodeling, characterized by progressive hypertrophy of myocytes and interstitial fibrosis, which in later stages involve progressive loss of myocytes through apoptosis, an exacerbated inflammatory response. The healing and the adverse remodeling of the infarcted ventricle ultimately underlie heart failure. This environment can lead to the secretion of certain factors into the circulation that are synthesized in various cell types in the heart, including cardiomyocytes, fibroblasts, smooth muscle, and vascular endothelial cells and other unknown factors. Image created with BioRender.com (Toronto, ON, Canada).
In this scenario, one of these secreted or leading factors can be components of RAAS, especially, AngII. This statement is based on several facts. On one hand, the activation of AngII/AT1R axis is generally associated with the pathophysiological appearance in HF, and it has also been established that RAAS is frequently altered in a variety of cancer types, which in turn is associated with a poor prognosis [13]. It should be considered that the pathological effects observed in these diseases are mainly associated with the AngII/ATR1 axis [45]. Thus, AT1R signaling increases aldosterone levels and blood pressure, induces vasoconstriction, cardiac hypertrophy, fibrosis, inflammation, and reactive oxygen species (ROS) production, while decreasing nitric oxide (NO) production, among other effects [12,42]. In the cancer scenario, AT1R activation by AngII favors cell proliferation, inhibits apoptosis, and promotes adhesion molecule expression, the interaction of monocytes with endothelial cells (EC), the infiltration of inflammatory cells, and the generation of pro-inflammatory cytokines, enabling the establishment of the inflammatory microenvironment, which is a pivotal state for the subsistence of neoplastic cells [47].
On another topic, it has previously been proposed that the initial immune response against a neoplasm is the result of the presence of an acute tissue injury that has generated a chronic infiltration of various myeloid cells, triggering a state of chronic inflammation in the tissue environment because the initial acute inflammatory response did not resolve [60]. Along the same lines, RAAS is a driver of tumorigenesis, linking with HF through immune and inflammatory responses. The involvement of immune cells both in the acute response after MI, the progression towards HF, and in the tumor microenvironment (TME) is a well-established notion. In the TME, immune cells intervene in various stages, mainly due to their infiltration into tumors and their differentiation into tumor-associated macrophages (TAM) [61], which are important components of the infiltration of most tumors and are derived mainly from circulating monocytes and which are attracted to the tumor by chemokines. In such a tumorigenic microenvironment, TAMs can stimulate tumor cell proliferation, promote angiogenesis, and favor invasion and metastasis [62]. In TME, they are frequently located surrounding blood vessels, where they secrete vascular endothelial growth factor (VEGF) and induce new blood vessel formation (angiogenesis). TAMs are the major immunoregulatory cells, and result in immune suppression in TME [63]. It should be noted that in the post-MI phase, monocytes predominate [32,57]. In MI, monocytes secrete angiogenic mediators, such as VEGF and the fibrogenic mediator transforming growth factor-beta (TGF-β), where their function is to promote the repair of infarcted tissue and cardiac fibrosis [57], so these HF-immune mediators can act as a substrate for the development of cancer cells by modulating a favorable microenvironment for its development (Figure 2).
En este escenario, uno de estos factores secretados o principales pueden ser componentes de RAAS, especialmente AngII. Esta declaración se basa en varios hechos. Por un lado, la activación del eje AngII / AT1R generalmente se asocia con el aspecto fisiopatológico en la IC, y también se ha establecido que el SRAA se encuentra frecuentemente alterado en una variedad de tipos de cáncer, lo que a su vez se asocia a un mal pronóstico [ 13 ]. Se debe considerar que los efectos patológicos observados en estas enfermedades están asociados principalmente con el eje AngII / ATR1 [ 45]. Así, la señalización de AT1R aumenta los niveles de aldosterona y la presión arterial, induce vasoconstricción, hipertrofia cardíaca, fibrosis, inflamación y producción de especies reactivas de oxígeno (ROS), mientras que disminuye la producción de óxido nítrico (NO), entre otros efectos [ 12 , 42 ]. En el escenario del cáncer, la activación de AT1R por AngII favorece la proliferación celular, inhibe la apoptosis y promueve la expresión de moléculas de adhesión, la interacción de los monocitos con las células endoteliales (CE), la infiltración de células inflamatorias y la generación de citocinas proinflamatorias, permitiendo la establecimiento del microambiente inflamatorio, que es un estado fundamental para la subsistencia de las células neoplásicas [ 47 ].
En otro tema, se ha propuesto previamente que la respuesta inmune inicial frente a una neoplasia es el resultado de la presencia de una lesión tisular aguda que ha generado una infiltración crónica de diversas células mieloides, desencadenando un estado de inflamación crónica en el entorno tisular debido a que la respuesta inflamatoria aguda inicial no se resolvió [ 60 ]. En la misma línea, RAAS es un impulsor de la tumorigénesis, que se vincula con la HF a través de respuestas inmunes e inflamatorias. La implicación de las células inmunitarias tanto en la respuesta aguda tras el IM, la progresión hacia la IC, como en el microambiente tumoral (TME) es una noción bien establecida. En la TME, las células inmunes intervienen en varias etapas, principalmente debido a su infiltración en los tumores y su diferenciación en macrófagos asociados a tumores (TAM) [61 ], que son componentes importantes de la infiltración de la mayoría de los tumores y se derivan principalmente de los monocitos circulantes y que son atraídos hacia el tumor por las quimiocinas. En un microambiente tumorigénico de este tipo, los TAM pueden estimular la proliferación de células tumorales, promover la angiogénesis y favorecer la invasión y la metástasis [ 62 ]. En la TME, con frecuencia se localizan alrededor de los vasos sanguíneos, donde secretan factor de crecimiento endotelial vascular (VEGF) e inducen la formación de nuevos vasos sanguíneos (angiogénesis). Las TAM son las principales células inmunorreguladoras y dan como resultado la supresión inmunitaria en la TME [ 63 ]. Cabe señalar que en la fase post-IM predominan los monocitos [ 32 , 57]. En el IM, los monocitos secretan mediadores angiogénicos, como el VEGF y el factor de crecimiento transformante del mediador fibrogénico-beta (TGF-β), donde su función es promover la reparación del tejido infartado y la fibrosis cardíaca [ 57 ], por lo que estos mediadores inmunes a la IC puede actuar como sustrato para el desarrollo de las células cancerosas modulando un microambiente favorable para su desarrollo ( Figura 2 ).
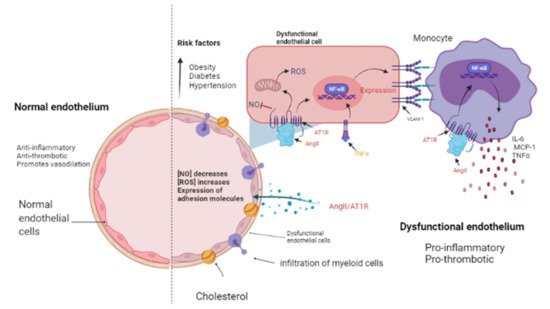
Figura 2. El endotelio en estado normal y patológico. El endotelio es una monocapa de células que cubre el interior de cada vaso mayor y menor. Un endotelio sano tiene propiedades antiinflamatorias y antitrombóticas y promueve la vasodilatación a través de la liberación de óxido nítrico (NO) ( lado izquierdo ). Los factores de riesgo cardiovascular como la obesidad, la diabetes y la hipertensión podrían promover un endotelio disfuncional ( lado derecho) que se caracteriza por una disminución en la liberación de NO así como un incremento en las especies reactivas de oxígeno (ROS) y una actividad proinflamatoria mediada por la señalización de AngII / AT1R, que activa NF-κB y, en consecuencia, la expresión de citocinas, quimiocinas y moléculas de adhesión: interleucina-6 (IL-6), proteína quimioatrayente de monocitos-1 (MCP-1) y molécula de adhesión de células vasculares-1 (VCAM-1) por células endoteliales. Luego, las células mieloides como los monocitos migran e infiltran hacia las paredes aórticas (donde se convierten en macrófagos) contribuyendo a la disfunción endotelial al producir factor de necrosis tumoral alfa (TNF-α), IL-6 y MCP-1. Imagen creada con BioRender.com, Toronto, ON, Canadá.
Figure 2. The endothelium in a normal and pathological state. The endothelium is a monolayer of cells that covers the interior of each major and minor vessel. A healthy endothelium has anti-inflammatory, anti-thrombotic properties and promotes vasodilation through nitric oxide (NO) release (left side). Cardiovascular risk factors such as obesity, diabetes and hypertension could promote a dysfunctional endothelium (right side) that is characterized by a decrease in NO release as well as an increment in reactive oxygen species (ROS) and a pro-inflammatory activity mediated by AngII/AT1R signaling, which activates NF-κB and, consequently, the expression of cytokines, chemokines, and adhesion molecules: interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1), and vascular cell adhesion molecule-1 (VCAM-1) by endothelial cells. Then, myeloid cells such as monocytes migrate and infiltrate towards the aortic walls (where they become macrophages) contributing to the endothelial dysfunction by producing tumor necrosis factor-alpha (TNF-α), IL-6, and MCP-1. Image created with BioRender.com, Toronto, ON, Canada.
Additionally, it was reported that systemic changes induced by the tumor influence the phenotype of circulating monocytes (such as the acquisition of immunosuppressive activity and a decreased responsiveness to inflammatory stimuli) before their infiltration into the tumor environment [61]. In particular, the inflammatory Ly6Chigh monocyte subset is efficiently recruited towards tumors and provides mediators that stimulate cancer-associated inflammation and angiogenesis [54,61]. Consistent with the above, Koelwyn et al. reported that MI accelerates breast cancer growth and cancer-specific mortality in mice and humans. In a murine model, there was an increase in the levels of circulating Ly6Chigh monocytes that were epigenetically reprogrammed in the spleen towards an immunosuppressive phenotype that was maintained in the tumor, as well as in the blood circulation, and additionally demonstrated that the depletion of these cells abolished MI-induced tumor growth [32]. AngII plays a relevant role in macrophage-mediated chronic inflammation by modulating this macrophage amplification program, since according to Retamozo et al. the overproduction of AngII increased macrophage progenitors in the spleen, allowing the extramedullary tissue to supply new macrophages associated with tumors throughout cancer progression in a tumor-bearing animal model. In contrast, blocking AngII production prevented the amplification of macrophage progenitors [54]. In this context, RAAS, and in particular AngII, could be a key point in the convergence of HF and cancer pathophysiology.
That is why it has been stated that chronic inflammation is a point of convergence between HF and cancer; because the former is characterized by chronic inflammation, it directly influences the risk of cancer development in patients with HF, since inflammation is an established component of carcinogenesis. Evidence suggests that chronic inflammation is responsible for up to 25% of all cancers [64]. On this occasion, we want to highlight the role of endothelial dysfunction as a pathophysiological substrate in the development of MI and HF, which in turn can generate an environment conducive to cancer progression.
The presence of cardiovascular risk factors, among them obesity, diabetes, and hypertension, directly affects the endothelium which is composed by EC. These cells line the inside of all major and minor vessels and serve as the first point of contact between the lumen and other tissues and regulate vascular tone, stiffness, inflammation, thrombotic potential in both health and illness (Figure 2). Cardiovascular risk factors mediate their detrimental effects on the vessel wall in part via enhanced activity of RAAS and increased release of vasoactive agents including Ang II as well as paracrine and circulating factors that regulate the generation and activity of endothelium-derived vasoactive and growth factors, adhesion molecules that mediate leucocyte-EC interaction, and blood coagulation regulators [65,66]. The endothelium in a healthy vasculature is anti-inflammatory, anti-thrombotic, and promotes vasodilation but, on the contrary, when the endothelium becomes dysfunctional, it is characterized by a pro-inflammatory and pro-thrombotic state [67,68] (Figure 2). It is worth noting that the exact mechanism by which a normally functioning endothelium becomes dysfunctional remain unknown. However, it has been reported that the endothelium is a prime site for the effects of cardiovascular risk factors; thus, endothelial function can be seen as an integrated index and sensitive measure of cardiovascular disease risk, since it reflects the cumulative contribution of various risk variables associated with inflammation and oxidative stress and given the similar pathological mechanisms that underpin cancer and cardiovascular disease [69]; thus, this is an elemental cellular component that can intermediate the transition between HF and cancer (Figure 2).
From a cancer perspective, dysfunctional ECs can promote pro-inflammatory signaling that is associated with characteristics that favor cancer progression, while in non-pathological conditions, it has been reported that ECs mitigate tumor invasiveness and metastasis [70]. Using in vitro models of dysfunctionally activated ECs, Franses et al. observed that resting EC constructs exhibited moderate inflammatory activity and could inhibit the proliferation and invasion of cancer cells. In contrast, “dysfunctional” ECs favored spontaneous metastasis in adjacent tumors through an aberrant expression of pro-inflammatory cytokines, extracellular matrix, alterations in the leukocyte adhesion process, increasing the expression of vascular cell adhesion molecule-1 (VCAM-1) and abnormal responses to oxidative stress, which are pathological stimuli present both in atherosclerotic lesions, precursors of MI and HF, as well as in the tumor environment (Figure 3) [66,70]. Molitor et al. provided evidence that the AngII/AT1R axis favors the migration and infiltration of myeloid cells towards the aortic walls, inducing endothelial dysfunction. Notably, AT1R blockade with telmisartan attenuated vascular infiltration of immune cells, reducing oxidative stress, and improved endothelial dysfunction [71]. Furthermore, in a subsequent study, they tested ACE inhibition in an HF model after MI, where they observed a decrease in systemic inflammation accompanied by a reduction in vascular infiltration of inflammatory myeloid cells and a decrease in the ROS levels nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) produced [55].
Además, se informó que los cambios sistémicos inducidos por el tumor influyen en el fenotipo de los monocitos circulantes (como la adquisición de actividad inmunosupresora y una menor capacidad de respuesta a los estímulos inflamatorios) antes de su infiltración en el entorno tumoral [ 61 ]. En particular, el subconjunto inflamatorio de monocitos altos Ly6C se recluta de manera eficiente hacia los tumores y proporciona mediadores que estimulan la inflamación asociada al cáncer y la angiogénesis [ 54 , 61 ]. De acuerdo con lo anterior, Koelwyn et al. informó que MI acelera el crecimiento del cáncer de mama y la mortalidad específica por cáncer en ratones y seres humanos. En un modelo murino, hubo un aumento en los niveles de Ly6C circulante alta monocitos que fueron reprogramados epigenéticamente en el bazo hacia un fenotipo inmunosupresor que se mantuvo en el tumor, así como en la circulación sanguínea, y además demostraron que el agotamiento de estas células abolió el crecimiento tumoral inducido por MI [ 32 ]. La AngII juega un papel relevante en la inflamación crónica mediada por macrófagos al modular este programa de amplificación de macrófagos, ya que según Retamozo et al. la sobreproducción de AngII aumentó los progenitores de macrófagos en el bazo, permitiendo que el tejido extramedular suministre nuevos macrófagos asociados con tumores a lo largo de la progresión del cáncer en un modelo animal portador de tumores. Por el contrario, el bloqueo de la producción de AngII impidió la amplificación de los progenitores de macrófagos [ 54]. En este contexto, RAAS, y en particular AngII, podría ser un punto clave en la convergencia de la fisiopatología de la IC y el cáncer.
Por eso se ha afirmado que la inflamación crónica es un punto de convergencia entre la IC y el cáncer; debido a que el primero se caracteriza por inflamación crónica, influye directamente en el riesgo de desarrollo de cáncer en pacientes con IC, ya que la inflamación es un componente establecido de la carcinogénesis. La evidencia sugiere que la inflamación crónica es responsable de hasta el 25% de todos los cánceres [ 64 ]. En esta ocasión queremos destacar el papel de la disfunción endotelial como sustrato fisiopatológico en el desarrollo de IM e IC, que a su vez pueden generar un entorno propicio para la progresión del cáncer.
La presencia de factores de riesgo cardiovascular, entre ellos obesidad, diabetes e hipertensión, afecta directamente al endotelio que está compuesto por CE. Estas células recubren el interior de todos los vasos mayores y menores y sirven como el primer punto de contacto entre la luz y otros tejidos y regulan el tono vascular, la rigidez, la inflamación y el potencial trombótico tanto en la salud como en la enfermedad ( Figura 2 ). Los factores de riesgo cardiovascular median sus efectos perjudiciales sobre la pared del vaso, en parte a través de una mayor actividad de RAAS y una mayor liberación de agentes vasoactivos, incluida Ang II, así como factores paracrinos y circulantes que regulan la generación y actividad de factores vasoactivos y de crecimiento derivados del endotelio, adhesión moléculas que median la interacción leucocito-EC y reguladores de la coagulación sanguínea [65 , 66 ]. El endotelio en una vasculatura sana es antiinflamatorio, antitrombótico y promueve la vasodilatación pero, por el contrario, cuando el endotelio se vuelve disfuncional, se caracteriza por un estado proinflamatorio y protrombótico [ 67 , 68 ] ( Figura 2). Vale la pena señalar que sigue sin conocerse el mecanismo exacto por el cual un endotelio que funciona normalmente se vuelve disfuncional. Sin embargo, se ha informado que el endotelio es un sitio principal para los efectos de los factores de riesgo cardiovascular; por lo tanto, la función endotelial puede verse como un índice integrado y una medida sensible del riesgo de enfermedad cardiovascular, ya que refleja la contribución acumulada de diversas variables de riesgo asociadas con la inflamación y el estrés oxidativo y dados los mecanismos patológicos similares que sustentan el cáncer y la enfermedad cardiovascular [ 69 ]. ; por tanto, este es un componente celular elemental que puede intermediar la transición entre la IC y el cáncer ( Figura 2 ).
Desde la perspectiva del cáncer, las CE disfuncionales pueden promover la señalización proinflamatoria que se asocia con características que favorecen la progresión del cáncer, mientras que en condiciones no patológicas, se ha informado que las CE mitigan la invasividad del tumor y la metástasis [ 70]. Utilizando modelos in vitro de CE activadas disfuncionalmente, Franses et al. observaron que las construcciones de EC en reposo exhibían una actividad inflamatoria moderada y podían inhibir la proliferación e invasión de células cancerosas. Por el contrario, las CE “disfuncionales” favorecieron la metástasis espontánea en tumores adyacentes a través de una expresión aberrante de citocinas proinflamatorias, matriz extracelular, alteraciones en el proceso de adhesión leucocitaria, aumentando la expresión de la molécula de adhesión celular vascular-1 (VCAM-1) y anormal respuestas al estrés oxidativo, que son estímulos patológicos presentes tanto en lesiones ateroscleróticas, precursoras de IM e IC, como en el entorno tumoral ( Figura 3 ) [ 66 , 70].]. Molitor y col. aportaron evidencias de que el eje AngII / AT1R favorece la migración e infiltración de células mieloides hacia las paredes aórticas, induciendo disfunción endotelial. En particular, el bloqueo de AT1R con telmisartán atenuó la infiltración vascular de las células inmunitarias, redujo el estrés oxidativo y mejoró la disfunción endotelial [ 71 ]. Además, en un estudio posterior, probaron la inhibición de la ECA en un modelo de HF después de MI, donde observaron una disminución de la inflamación sistémica acompañada de una reducción de la infiltración vascular de células mieloides inflamatorias y una disminución de los niveles de ROS nicotinamida adenina dinucleótido fosfato oxidasa ( NADPH oxidasa) producido [ 55 ].
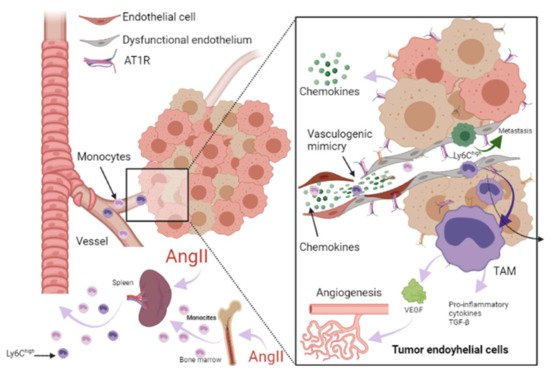
Figura 3. Papel de RAAS en el microambiente tumoral. Las células inmunes pueden infiltrar tumores y diferenciarse en macrófagos asociados a tumores (TAM) derivados principalmente de monocitos circulantes y son atraídas hacia el tumor por quimiocinas. Los TAM pueden estimular la proliferación de células tumorales, la angiogénesis, la invasión y la metástasis. Además, el microambiente tumoral (TME) puede influir en el fenotipo de los monocitos circulantes como el Ly6C alto. subconjunto de monocitos, dándoles una actividad inmunosupresora y una menor capacidad de respuesta a los estímulos inflamatorios antes de su infiltración en TME. La angiotensina-II (AngII) juega un papel relevante en la inflamación crónica mediada por macrófagos, aumentando los progenitores de macrófagos y proporcionando TAM. Adicionalmente, las células endoteliales (CE) pueden promover la señalización proinflamatoria, favoreciendo la metástasis espontánea en tumores adyacentes a través de una expresión aberrante de citocinas proinflamatorias, matriz extracelular, alteraciones en el proceso de adhesión leucocitaria, aumento de la adhesión celular vascular molécula-1 (VCAM- 1) y respuestas anormales al estrés oxidativo. En las CE, la señalización de AngII / AT1R genera una respuesta proangiogénica mediada por el factor de crecimiento endotelial vascular (VEGF). La señalización de AngII activa TNF-α y NF-κB y regula al alza las quimiocinas endoteliales proinflamatorias. Se ha informado que AngII puede promover la expresión de VCAM-1 y mejorar la adhesión, el crecimiento, la angiogénesis y el microambiente inflamatorio a través de AT1R en el carcinoma hepatocelular. Se ha informado que la angiogénesis promueve la metástasis de células tumorales. Imagen creada con BioRender.com, Toronto, ON, Canadá.
Figure 3. Role of RAAS in the tumor microenvironment. Immune cells can infiltrate tumors and differentiate into tumor-associated macrophages (TAM) derived mainly from circulating monocytes and are attracted to the tumor by chemokines. TAMs can stimulate tumor cell proliferation, angiogenesis, invasion and metastasis. Additionally, tumor microenvironment (TME) can influence the phenotype of circulating monocytes such as the Ly6Chigh monocyte subset, giving them an immunosuppressive activity and a decreased responsiveness to inflammatory stimuli before their infiltration intoTME. Angiotensin-II (AngII) plays a relevant role in macrophage-mediated chronic inflammation, increasing macrophage progenitors and supplying of TAMs. Additionally, endothelial cells (EC) can promote pro-inflammatory signaling, favoring spontaneous metastasis in adjacent tumors through an aberrant expression of pro-inflammatory cytokines, extracellular matrix, alterations in the leukocyte adhesion process, increasing vascular cell adhesion molecule-1 (VCAM-1) and abnormal responses to oxidative stress. In ECs, AngII/AT1R signaling generates a pro-angiogenic response mediated by vascular endothelial growth factor (VEGF). AngII signaling activates TNF-α and NF-κB and upregulates pro-inflammatory endothelial chemokines. AngII has been reported to be able to promote VCAM-1 expression and enhance adhesion, growth, angiogenesis, and the inflammatory microenvironment through AT1R in hepatocellular carcinoma. It has been reported that angiogenesis promotes tumor cell metastasis. Image created with BioRender.com, Toronto, ON, Canada.
ECs are essential in the tumor microenvironment, as they can also express and use components of the RAAS signaling pathway to promote tumor growth, enhance angiogenesis, and promote metastasis [13,72]. It should be noted that adhesion molecules play a crucial role in these processes, since they allow the union and transendothelial migration, in this case, of tumor cells or TAMs. AngII signaling activates TNF-α and NF-κB and upregulates pro-inflammatory endothelial chemokines [66,73]. AngII has been reported to be able to promote VCAM-1 expression and enhance adhesion, growth, angiogenesis, and the inflammatory microenvironment through AT1R in hepatocellular carcinoma [74]. During HF, an increase in VCAM-1 expression was observed in response to AngII stimulation, and this is associated with endothelial dysfunction as well [71,75].
Angiogenesis is involved in myocardial healing. It has been reported to promote tumor progression, depending first on supplying oxygen and nutrients and later, generating a pathway for its metastasis [14]. In ECs, AT1R-mediated AngII generates a pro-angiogenic response mediated by VEGF, a crucial stimulator of pathological vessel formation (Figure 3) [76]. Therefore, this represents another mechanism in which HF could influence the environment that leads to cancer progression. Thus, the study of endothelial dysfunction mechanisms is crucial to preventing the recurrence of serious secondary events in patients who have suffered HF, including the development of cancer.
Las CE son esenciales en el microambiente tumoral, ya que también pueden expresar y utilizar componentes de la vía de señalización RAAS para promover el crecimiento tumoral, mejorar la angiogénesis y promover la metástasis [ 13 , 72 ]. Cabe destacar que las moléculas de adhesión juegan un papel crucial en estos procesos, ya que permiten la unión y migración transendotelial, en este caso, de células tumorales o TAM. La señalización de AngII activa TNF-α y NF-κB y regula al alza las quimiocinas endoteliales proinflamatorias [ 66 , 73 ]. Se ha informado que la AngII puede promover la expresión de VCAM-1 y mejorar la adhesión, el crecimiento, la angiogénesis y el microambiente inflamatorio a través de AT1R en el carcinoma hepatocelular [ 74]. Durante la IC, se observó un aumento en la expresión de VCAM-1 en respuesta a la estimulación de AngII, y esto también se asocia con disfunción endotelial [ 71 , 75 ].
La angiogénesis está involucrada en la curación del miocardio. Se ha informado que promueve la progresión del tumor, dependiendo primero del suministro de oxígeno y nutrientes y luego, de generar una vía para su metástasis [ 14 ]. En las CE, la AngII mediada por AT1R genera una respuesta proangiogénica mediada por VEGF, un estimulador crucial de la formación de vasos patológicos ( Figura 3 ) [ 76 ]. Por tanto, esto representa otro mecanismo en el que la IC podría influir en el entorno que conduce a la progresión del cáncer. Así, el estudio de los mecanismos de disfunción endotelial es crucial para prevenir la recurrencia de eventos secundarios graves en pacientes que han sufrido IC, incluido el desarrollo de cáncer.