La insuficiencia cardíaca (IC) se asocia con muchas comorbilidades y, entre ellas, el cáncer se ha destacado como contribuyente de muerte en estos pacientes. Los pacientes con IC tienen un mayor riesgo de cáncer en comparación con la población general. Estas dos patologías comparten una variedad de factores de riesgo cardiovascular, así como mecanismos fisiopatológicos. Se han propuesto dos hipótesis sobre los mecanismos fisiopatológicos compartidos: un entorno inflamatorio constante y una activación neurohumoral crónica.La respuesta neurohumoral incluye el sistema renina-angiotensina-aldosterona (RAAS), que puede ser estimulado crónicamente, lo que representa un sello distintivo de la IC, y también se ha establecido que el RAAS se altera con frecuencia en una variedad de tipos de cáncer , lo que se asocia con una mala pronóstico.
1. Sistema renina-angiotensina-aldosterona que vincula la insuficiencia cardíaca isquémica y el cáncer
Hay dos vías principales en el RAAS: vías clásicas y no clásicas. En el RAAS clásico, el péptido efector es la angiotensina-II (AngII), que se produce a partir de su precursor hepático, el angiotensinógeno, que es catabolizado por la enzima renina, dando a su vez a la angiotensina-I (AngI ), que es un sustrato. para la enzima convertidora de angiotensina (ECA) que produce angiotensina-II. Los efectos funcionales de AngII en el RAAS clásicos están mediados en gran medida por el receptor de angiotensina II de tipo 1 (AT1R) y el receptor de tipo 2 (AT2R) [ 12 , 35 ]. La activación de AT1R aumenta la aldosterona, un actor importante en la regulación del equilibrio electrolítico [ 36], pero también la activación de AT1R tiene muchos otros efectos (descritos más adelante). La señalización mediada por el AT2R se asocia con funciones antifibróticas e incluso con efectos antiinflamatorios en la IC [ 37 , 38 , 39 ], mientras que en el cáncer este eje tiene efectos antiproliferativos, antiangiogénicos y proapoptóticos [ 40 , 41 ]. Sin embargo, también hay informes contradictorios que sugirieron posibles diferencias específicas del tipo de tumor [ 36 , 40 ].
En el RAAS no clásico, el homólogo de ACE, la enzima convertidora de angiotensina 2 (ACE2) escinde AngI en un nonapéptido, Ang 1-9 y AngII en un heptapéptido, Ang 1-7. Además, AngII también se puede convertir en Ang 2-8 (AngIII) por la aminopeptidasa A, y ejerce sus efectos al unirse a AT1R. La aminopeptidasa N convierte AngIII en Ang 3-8 (AngIV) y puede actuar a través del receptor de angiotensina 4 (AT4R) [ 12 ]. Ang 1-9 puede activar AT2R, y Ang 1-7 puede unirse al receptor del protooncogén Mas (MasR). Curiosamente, se ha demostrado que cada uno de estos componentes contrarresta las acciones del RAAS clásico [ 12 , 42]. Se ha demostrado que la señalización mediada por el eje ACE2 / Ang 1-7 / MasR tiene un papel protector en el desarrollo de la remodelación del miocardio después de un IM en un modelo animal [ 43 ], pero también se asocia con efectos antifibróticos y antiinflamatorios [ 44 , 45 ]. Además, la señalización de AngIV / AT4R tiene un papel cardioprotector, actuando como contraparte de la inflamación mediada por Ang II y la fibrosis miocárdica en el modelo de rata [ 46 ]. En el cáncer, se ha documentado que MasR reduce la angiogénesis anormal, la inflamación y la proliferación celular mediante la disminución local de los niveles de Ang II o el bloqueo del receptor AT1 asociado con altas concentraciones de Ang (1-7) en el sitio del tumor [ 47]. Aun así, dado que el AT1R sigue siendo crucial en la mediación de los efectos fisiológicos y fisiopatológicos de la AngII [ 48 ], en esta revisión nos vamos a centrar en el eje clásico AT1R / AngII RAAS.
La sobreproducción de AngII está relacionada con el desarrollo de enfermedades crónicas; de hecho, una activación crónica del RAAS es un sello distintivo de la IC, especialmente marcada por un aumento sistémico en los niveles de AngII [ 10 , 49 ], y para comprender mejor cómo el RAAS está implicado en ambas enfermedades, primero debemos considerar los implicados. fisiopatología de MI a HF y luego al cáncer.
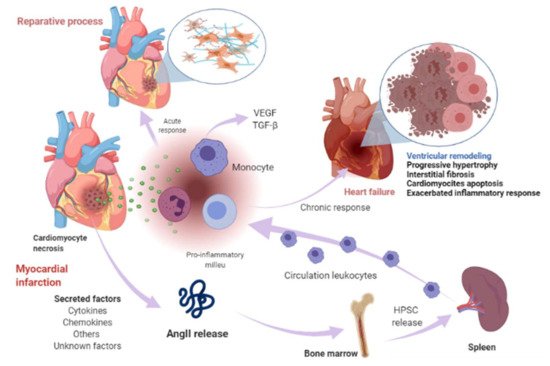
La necrosis de cardiomiocitos en el miocardio infartado activa la respuesta inmune innata, desencadenando una respuesta inflamatoria. La liberación de señales de peligro de las células moribundas induce la secreción de citocinas, como quimiocinas y moléculas de adhesión, para permitir el reclutamiento e infiltración de leucocitos, principalmente monocitos, en el área infartada, donde ejercen una respuesta "reparadora", fagocitando los restos celulares. , mientras estimula las vías de reparación mediante la secreción de citocinas proinflamatorias. Para suministrar la cantidad adecuada de células inmunitarias, se produce una liberación de células madre y progenitores hematopoyéticos de los nichos de la médula ósea; estas células luego migran al bazo y, en última instancia, aumentan la producción de células inmunes, que a su vez media una respuesta inflamatoria eficaz ( Figura 1 ) [50 , 51 ]. La modulación de la inflamación en esta fase de reparación incluye la activación y curación de fibroblastos mediada por la respuesta neurohumoral. El SRAA, que forma parte de la respuesta neurohumoral, se activa por hipoperfusión renal y activación simpática como mecanismos compensatorios después de una lesión miocárdica [ 11 ]. En estos procesos, RAAS participa activamente principalmente a través del péptido efector AngII. De hecho, se ha informado que varios componentes de RAAS, incluidos angiotensinógeno, AngII, ACE, AT1R y AT2R, se expresan en una variedad de células inmunes [ 52 ], así como en células de la médula ósea [ 53]. Poco después de la lesión miocárdica, se produce un aumento en la concentración de AngII, que induce la acumulación, diferenciación y salida de células madre / precursoras hematopoyéticas (HPSC) de la médula ósea para contribuir a la mielopoyesis esplénica, suministrando hasta el 50% de los leucocitos a el área infartada [ 54 ], ya través de la fosforilación del factor nuclear kappa B (NF-kB), la unión de AngII a su receptor AT1R induce una respuesta proinflamatoria mediada por el factor de necrosis tumoral alfa (TNF-α) o interleucina-1 beta (IL1ß), que a su vez son impulsoras de la inflamación [ 55]. Sin embargo, cuando la respuesta neurohumoral se vuelve crónica, conduce a una pérdida excesiva de cardiomiocitos, una respuesta inflamatoria exacerbada y la curación y remodelación adversa del ventrículo infartado, que finalmente subyace a la IC [ 56 , 57 , 58 ]. Se ha propuesto que este entorno disfuncional desencadena la secreción de varios factores en la circulación que pueden sintetizarse en varios tipos de células que rodean el corazón y sus propios componentes celulares, incluidos cardiomiocitos, fibroblastos, músculo liso (progenitores aórticos o derivados de la sangre) y células endoteliales vasculares ( Figura 1 ) [ 33 , 59 ].
Figura 1. Infarto de miocardio y eventos relacionados con insuficiencia cardíaca. Poco después de la lesión miocárdica, se produce un aumento de la concentración de AngII e induce la acumulación, diferenciación y salida de células madre / precursoras hematopoyéticas (HPSC) de la médula ósea para contribuir a la mielopoyesis esplénica para irrigar el área infartada de las células inmunitarias. La necrosis de cardiomiocitos libera señales de peligro e induce la secreción de citocinas, quimiocinas y moléculas de adhesión para permitir el reclutamiento e infiltración de leucocitos (principalmente monocitos) en el área infartada. Los monocitos ejercen una respuesta reparadora, fagocitosan los restos celulares, mientras que estimulan las vías de reparación secretando citocinas proinflamatorias a través de la unión de la angiotensina-II (AngII) al receptor de angiotensina-II tipo 1 (AT1R), que induce la fosforilación del factor nuclear kappa B (NF-kB). Esto induce una respuesta proinflamatoria mediada por el factor de necrosis tumoral alfa (TNF-α) o interleucina-1 beta (IL1ß) e impulsa la inflamación. La modulación de la inflamación en esta fase de reparación incluye la activación y curación de fibroblastos mediada en parte por el sistema renina-angiotensina-aldosterona (RAAS). Cuando esta respuesta se vuelve crónica, conduce a un proceso patológico llamado remodelado ventricular, caracterizado por hipertrofia progresiva de miocitos y fibrosis intersticial, que en etapas posteriores implica pérdida progresiva de miocitos por apoptosis, una respuesta inflamatoria exacerbada. La curación y la remodelación adversa del ventrículo infartado subyacen en última instancia a la insuficiencia cardíaca. Este entorno puede conducir a la secreción de ciertos factores en la circulación que se sintetizan en varios tipos de células en el corazón, incluidos cardiomiocitos, fibroblastos, músculo liso y células endoteliales vasculares y otros factores desconocidos. Imagen creada con BioRender.com (Toronto, ON, Canadá).
En este escenario, uno de estos factores secretados o principales pueden ser componentes de RAAS, especialmente AngII. Esta declaración se basa en varios hechos. Por un lado, la activación del eje AngII / AT1R generalmente se asocia con el aspecto fisiopatológico en la IC, y también se ha establecido que el SRAA se encuentra frecuentemente alterado en una variedad de tipos de cáncer, lo que a su vez se asocia a un mal pronóstico [ 13 ]. Se debe considerar que los efectos patológicos observados en estas enfermedades están asociados principalmente con el eje AngII / ATR1 [ 45]. Así, la señalización de AT1R aumenta los niveles de aldosterona y la presión arterial, induce vasoconstricción, hipertrofia cardíaca, fibrosis, inflamación y producción de especies reactivas de oxígeno (ROS), mientras que disminuye la producción de óxido nítrico (NO), entre otros efectos [ 12 , 42 ]. En el escenario del cáncer, la activación de AT1R por AngII favorece la proliferación celular, inhibe la apoptosis y promueve la expresión de moléculas de adhesión, la interacción de los monocitos con las células endoteliales (CE), la infiltración de células inflamatorias y la generación de citocinas proinflamatorias, permitiendo la establecimiento del microambiente inflamatorio, que es un estado fundamental para la subsistencia de las células neoplásicas [ 47 ].
En otro tema, se ha propuesto previamente que la respuesta inmune inicial frente a una neoplasia es el resultado de la presencia de una lesión tisular aguda que ha generado una infiltración crónica de diversas células mieloides, desencadenando un estado de inflamación crónica en el entorno tisular debido a que la respuesta inflamatoria aguda inicial no se resolvió [ 60 ]. En la misma línea, RAAS es un impulsor de la tumorigénesis, que se vincula con la HF a través de respuestas inmunes e inflamatorias. La implicación de las células inmunitarias tanto en la respuesta aguda tras el IM, la progresión hacia la IC, como en el microambiente tumoral (TME) es una noción bien establecida. En la TME, las células inmunes intervienen en varias etapas, principalmente debido a su infiltración en los tumores y su diferenciación en macrófagos asociados a tumores (TAM) [61 ], que son componentes importantes de la infiltración de la mayoría de los tumores y se derivan principalmente de los monocitos circulantes y que son atraídos hacia el tumor por las quimiocinas. En un microambiente tumorigénico de este tipo, los TAM pueden estimular la proliferación de células tumorales, promover la angiogénesis y favorecer la invasión y la metástasis [ 62 ]. En la TME, con frecuencia se localizan alrededor de los vasos sanguíneos, donde secretan factor de crecimiento endotelial vascular (VEGF) e inducen la formación de nuevos vasos sanguíneos (angiogénesis). Las TAM son las principales células inmunorreguladoras y dan como resultado la supresión inmunitaria en la TME [ 63 ]. Cabe señalar que en la fase post-IM predominan los monocitos [ 32 , 57]. En el IM, los monocitos secretan mediadores angiogénicos, como el VEGF y el factor de crecimiento transformante del mediador fibrogénico-beta (TGF-β), donde su función es promover la reparación del tejido infartado y la fibrosis cardíaca [ 57 ], por lo que estos mediadores inmunes a la IC puede actuar como sustrato para el desarrollo de las células cancerosas modulando un microambiente favorable para su desarrollo ( Figura 2 ).
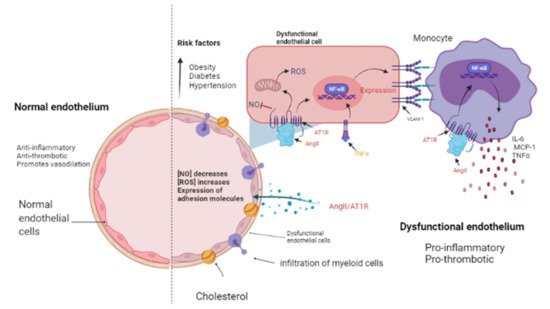
Figura 2. El endotelio en estado normal y patológico. El endotelio es una monocapa de células que cubre el interior de cada vaso mayor y menor. Un endotelio sano tiene propiedades antiinflamatorias y antitrombóticas y promueve la vasodilatación a través de la liberación de óxido nítrico (NO) ( lado izquierdo ). Los factores de riesgo cardiovascular como la obesidad, la diabetes y la hipertensión podrían promover un endotelio disfuncional ( lado derecho) que se caracteriza por una disminución en la liberación de NO así como un incremento en las especies reactivas de oxígeno (ROS) y una actividad proinflamatoria mediada por la señalización de AngII / AT1R, que activa NF-κB y, en consecuencia, la expresión de citocinas, quimiocinas y moléculas de adhesión: interleucina-6 (IL-6), proteína quimioatrayente de monocitos-1 (MCP-1) y molécula de adhesión de células vasculares-1 (VCAM-1) por células endoteliales. Luego, las células mieloides como los monocitos migran e infiltran hacia las paredes aórticas (donde se convierten en macrófagos) contribuyendo a la disfunción endotelial al producir factor de necrosis tumoral alfa (TNF-α), IL-6 y MCP-1. Imagen creada con BioRender.com, Toronto, ON, Canadá.
Además, se informó que los cambios sistémicos inducidos por el tumor influyen en el fenotipo de los monocitos circulantes (como la adquisición de actividad inmunosupresora y una menor capacidad de respuesta a los estímulos inflamatorios) antes de su infiltración en el entorno tumoral [ 61 ]. En particular, el subconjunto inflamatorio de monocitos altos Ly6C se recluta de manera eficiente hacia los tumores y proporciona mediadores que estimulan la inflamación asociada al cáncer y la angiogénesis [ 54 , 61 ]. De acuerdo con lo anterior, Koelwyn et al. informó que MI acelera el crecimiento del cáncer de mama y la mortalidad específica por cáncer en ratones y seres humanos. En un modelo murino, hubo un aumento en los niveles de Ly6C circulante alta monocitos que fueron reprogramados epigenéticamente en el bazo hacia un fenotipo inmunosupresor que se mantuvo en el tumor, así como en la circulación sanguínea, y además demostraron que el agotamiento de estas células abolió el crecimiento tumoral inducido por MI [ 32 ]. La AngII juega un papel relevante en la inflamación crónica mediada por macrófagos al modular este programa de amplificación de macrófagos, ya que según Retamozo et al. la sobreproducción de AngII aumentó los progenitores de macrófagos en el bazo, permitiendo que el tejido extramedular suministre nuevos macrófagos asociados con tumores a lo largo de la progresión del cáncer en un modelo animal portador de tumores. Por el contrario, el bloqueo de la producción de AngII impidió la amplificación de los progenitores de macrófagos [ 54]. En este contexto, RAAS, y en particular AngII, podría ser un punto clave en la convergencia de la fisiopatología de la IC y el cáncer.
Por eso se ha afirmado que la inflamación crónica es un punto de convergencia entre la IC y el cáncer; debido a que el primero se caracteriza por inflamación crónica, influye directamente en el riesgo de desarrollo de cáncer en pacientes con IC, ya que la inflamación es un componente establecido de la carcinogénesis. La evidencia sugiere que la inflamación crónica es responsable de hasta el 25% de todos los cánceres [ 64 ]. En esta ocasión queremos destacar el papel de la disfunción endotelial como sustrato fisiopatológico en el desarrollo de IM e IC, que a su vez pueden generar un entorno propicio para la progresión del cáncer.
La presencia de factores de riesgo cardiovascular, entre ellos obesidad, diabetes e hipertensión, afecta directamente al endotelio que está compuesto por CE. Estas células recubren el interior de todos los vasos mayores y menores y sirven como el primer punto de contacto entre la luz y otros tejidos y regulan el tono vascular, la rigidez, la inflamación y el potencial trombótico tanto en la salud como en la enfermedad ( Figura 2 ). Los factores de riesgo cardiovascular median sus efectos perjudiciales sobre la pared del vaso, en parte a través de una mayor actividad de RAAS y una mayor liberación de agentes vasoactivos, incluida Ang II, así como factores paracrinos y circulantes que regulan la generación y actividad de factores vasoactivos y de crecimiento derivados del endotelio, adhesión moléculas que median la interacción leucocito-EC y reguladores de la coagulación sanguínea [65 , 66 ]. El endotelio en una vasculatura sana es antiinflamatorio, antitrombótico y promueve la vasodilatación pero, por el contrario, cuando el endotelio se vuelve disfuncional, se caracteriza por un estado proinflamatorio y protrombótico [ 67 , 68 ] ( Figura 2). Vale la pena señalar que sigue sin conocerse el mecanismo exacto por el cual un endotelio que funciona normalmente se vuelve disfuncional. Sin embargo, se ha informado que el endotelio es un sitio principal para los efectos de los factores de riesgo cardiovascular; por lo tanto, la función endotelial puede verse como un índice integrado y una medida sensible del riesgo de enfermedad cardiovascular, ya que refleja la contribución acumulada de diversas variables de riesgo asociadas con la inflamación y el estrés oxidativo y dados los mecanismos patológicos similares que sustentan el cáncer y la enfermedad cardiovascular [ 69 ]. ; por tanto, este es un componente celular elemental que puede intermediar la transición entre la IC y el cáncer ( Figura 2 ).
Desde la perspectiva del cáncer, las CE disfuncionales pueden promover la señalización proinflamatoria que se asocia con características que favorecen la progresión del cáncer, mientras que en condiciones no patológicas, se ha informado que las CE mitigan la invasividad del tumor y la metástasis [ 70]. Utilizando modelos in vitro de CE activadas disfuncionalmente, Franses et al. observaron que las construcciones de EC en reposo exhibían una actividad inflamatoria moderada y podían inhibir la proliferación e invasión de células cancerosas. Por el contrario, las CE “disfuncionales” favorecieron la metástasis espontánea en tumores adyacentes a través de una expresión aberrante de citocinas proinflamatorias, matriz extracelular, alteraciones en el proceso de adhesión leucocitaria, aumentando la expresión de la molécula de adhesión celular vascular-1 (VCAM-1) y anormal respuestas al estrés oxidativo, que son estímulos patológicos presentes tanto en lesiones ateroscleróticas, precursoras de IM e IC, como en el entorno tumoral ( Figura 3 ) [ 66 , 70].]. Molitor y col. aportaron evidencias de que el eje AngII / AT1R favorece la migración e infiltración de células mieloides hacia las paredes aórticas, induciendo disfunción endotelial. En particular, el bloqueo de AT1R con telmisartán atenuó la infiltración vascular de las células inmunitarias, redujo el estrés oxidativo y mejoró la disfunción endotelial [ 71 ]. Además, en un estudio posterior, probaron la inhibición de la ECA en un modelo de HF después de MI, donde observaron una disminución de la inflamación sistémica acompañada de una reducción de la infiltración vascular de células mieloides inflamatorias y una disminución de los niveles de ROS nicotinamida adenina dinucleótido fosfato oxidasa ( NADPH oxidasa) producido [ 55 ].
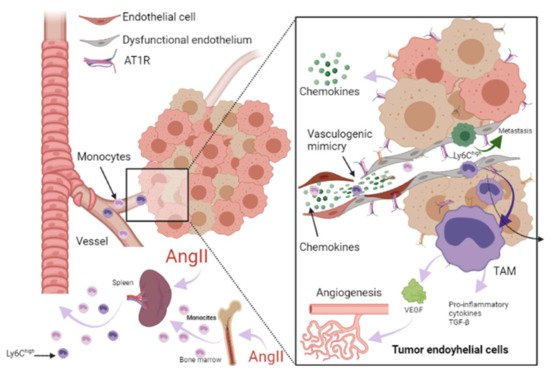
Figura 3. Papel de RAAS en el microambiente tumoral. Las células inmunes pueden infiltrar tumores y diferenciarse en macrófagos asociados a tumores (TAM) derivados principalmente de monocitos circulantes y son atraídas hacia el tumor por quimiocinas. Los TAM pueden estimular la proliferación de células tumorales, la angiogénesis, la invasión y la metástasis. Además, el microambiente tumoral (TME) puede influir en el fenotipo de los monocitos circulantes como el Ly6C alto. subconjunto de monocitos, dándoles una actividad inmunosupresora y una menor capacidad de respuesta a los estímulos inflamatorios antes de su infiltración en TME. La angiotensina-II (AngII) juega un papel relevante en la inflamación crónica mediada por macrófagos, aumentando los progenitores de macrófagos y proporcionando TAM. Adicionalmente, las células endoteliales (CE) pueden promover la señalización proinflamatoria, favoreciendo la metástasis espontánea en tumores adyacentes a través de una expresión aberrante de citocinas proinflamatorias, matriz extracelular, alteraciones en el proceso de adhesión leucocitaria, aumento de la adhesión celular vascular molécula-1 (VCAM- 1) y respuestas anormales al estrés oxidativo. En las CE, la señalización de AngII / AT1R genera una respuesta proangiogénica mediada por el factor de crecimiento endotelial vascular (VEGF). La señalización de AngII activa TNF-α y NF-κB y regula al alza las quimiocinas endoteliales proinflamatorias. Se ha informado que AngII puede promover la expresión de VCAM-1 y mejorar la adhesión, el crecimiento, la angiogénesis y el microambiente inflamatorio a través de AT1R en el carcinoma hepatocelular. Se ha informado que la angiogénesis promueve la metástasis de células tumorales. Imagen creada con BioRender.com, Toronto, ON, Canadá.
Las CE son esenciales en el microambiente tumoral, ya que también pueden expresar y utilizar componentes de la vía de señalización RAAS para promover el crecimiento tumoral, mejorar la angiogénesis y promover la metástasis [ 13 , 72 ]. Cabe destacar que las moléculas de adhesión juegan un papel crucial en estos procesos, ya que permiten la unión y migración transendotelial, en este caso, de células tumorales o TAM. La señalización de AngII activa TNF-α y NF-κB y regula al alza las quimiocinas endoteliales proinflamatorias [ 66 , 73 ]. Se ha informado que la AngII puede promover la expresión de VCAM-1 y mejorar la adhesión, el crecimiento, la angiogénesis y el microambiente inflamatorio a través de AT1R en el carcinoma hepatocelular [ 74]. Durante la IC, se observó un aumento en la expresión de VCAM-1 en respuesta a la estimulación de AngII, y esto también se asocia con disfunción endotelial [ 71 , 75 ].
La angiogénesis está involucrada en la curación del miocardio. Se ha informado que promueve la progresión del tumor, dependiendo primero del suministro de oxígeno y nutrientes y luego, de generar una vía para su metástasis [ 14 ]. En las CE, la AngII mediada por AT1R genera una respuesta proangiogénica mediada por VEGF, un estimulador crucial de la formación de vasos patológicos ( Figura 3 ) [ 76 ]. Por tanto, esto representa otro mecanismo en el que la IC podría influir en el entorno que conduce a la progresión del cáncer. Así, el estudio de los mecanismos de disfunción endotelial es crucial para prevenir la recurrencia de eventos secundarios graves en pacientes que han sufrido IC, incluido el desarrollo de cáncer.
2. Papel de la farmacoterapia con inhibidores del RAAS en la insuficiencia cardíaca y el cáncer
Los inhibidores de la enzima convertidora de angiotensina (IECA) y los bloqueadores de los receptores de angiotensina (ARA) forman parte del tratamiento farmacológico básico tras el IM (además de la reperfusión), y se ha informado que su administración reduce la mortalidad a corto y largo plazo además de reducir el riesgo de HF [ 77 , 78 , 79 ]. Además, datos recientes sugieren que estos fármacos también poseen características antiinflamatorias y anticancerígenas [ 80 , 81 ], lo que refuerza el papel convergente del sistema RAAS en la IC y el cáncer, porque sus inhibidores moleculares juegan un papel importante en el desarrollo, la migración y la recurrencia y resistencia a los fármacos antineoplásicos [ 81 ].
Para denotar el papel que juega el RAAS en la IC y el cáncer, las propiedades terapéuticas que poseen los inhibidores del RAAS en la IC ( Tabla 1 ) y los hallazgos que estos inhibidores han mostrado en la investigación preclínica del cáncer ( Tabla 2 ).
Tabla 1. Papel de los inhibidores de RAAS en la insuficiencia cardíaca.
| Inhibidor de RAAS |
Observaciones |
| Inhibidores de la enzima convertidora de angiotensina (IECA) |
| Captopril |
La administración a largo plazo se asoció con una mejora de la supervivencia y una reducción de la morbilidad y la mortalidad debidas a eventos cardiovasculares importantes en pacientes con disfunción asintomática del ventrículo izquierdo (VI) después de un infarto de miocardio (IM) [ 82 ]. |
| Enalapril |
Mayor tiempo de ejercicio y fracción de eyección del ventrículo izquierdo (FEVI) [ 83 ]. |
| Perindopril |
Aumentó la distancia de caminata de 6 min, pero no disminuyó la mortalidad [ 84 ]. Después de un año de tratamiento, se redujo la remodelación progresiva del VI, pero no se asoció con mejores resultados clínicos [ 85 ]. |
| Ramipril |
La administración a pacientes con evidencia clínica de insuficiencia cardíaca (IC) transitoria o continua después de un infarto de miocardio resultó en una reducción sustancial de la muerte prematura por todas las causas [ 86 ]. |
| Trandolapril |
El tratamiento a largo plazo en pacientes con función del VI reducida poco después de un IM redujo significativamente el riesgo de mortalidad global, mortalidad por causas cardiovasculares, muerte súbita y desarrollo de IC grave [ 87 ]. |
| Bloqueadores de los receptores de angiotensina II tipo 1 (BRA) |
| Telmisartan |
El telmisartán fue bien tolerado en pacientes que no pudieron tolerar los IECA. Aunque el fármaco no tuvo un efecto significativo sobre las hospitalizaciones por IC, redujo modestamente el riesgo del resultado combinado de muerte cardiovascular, infarto de miocardio o accidente cerebrovascular [ 88 ]. |
| Candesartán |
Disminuyó ligeramente las hospitalizaciones, pero no disminuyó la mortalidad [ 89 ]. Reducción de la mortalidad cardiovascular y los ingresos hospitalarios por empeoramiento de la IC crónica. Los pacientes con fracción de eyección reducida fueron los más beneficiados [ 90 ]. |
| Losartán |
Reducción de la tasa de muerte o ingreso por IC en pacientes con IC, reducción de la FEVI e intolerancia a los IECA [ 91 ]. |
| Valsartán |
En pacientes con IM asociado con IC y / o disfunción del VI, la administración de valsartán en el período inmediatamente posterior al IM demostró la misma eficacia que captopril [ 92 , 93 ]. |
| Antagonistas de la aldosterona |
| Espironolactona |
Prevención de la fibrosis y remodelación del VI después de un IM [ 94 ] |
Tabla 2. Papel del inhibidor de RAA en la investigación preclínica del cáncer.
| Inhibidor de RAAS |
Recomendaciones |
| Inhibidores de la enzima convertidora de angiotensina (IECA) |
| Captopril |
Inhibe el crecimiento tumoral en un modelo de cáncer gástrico y suprime la angiogénesis del tumor al disminuir la expresión del factor de crecimiento endotelial vascular (VEGF) y la metaloproteinasa de matriz (MMP) -7 en un modelo de ratón con cáncer gástrico humano [ 99 ]. Atenúa la migración celular en un modelo de cáncer de mama [ 100 ]. Inhibe el crecimiento celular, disminuye la expresión de c-myc y aumenta la apoptosis en las líneas celulares leucémicas [ 101 ]. |
| Enalapril |
Inhibe la progresión tumoral y reduce el número de macrófagos asociados a tumores (TAM) [ 54 ]. |
| Perindopril |
Puede inhibir el crecimiento tumoral en el modelo de cáncer gástrico y suprimir la angiogénesis del tumor al disminuir la expresión de VEGF y MMP-7 en un modelo de ratón con cáncer gástrico humano [ 99 ]. |
| Ramipril |
Disminuye la inflamación sistémica [ 55 ]. |
| Trandolapril |
Inhibe el crecimiento celular, disminuye la expresión de c-myc y aumenta la apoptosis en líneas celulares leucémicas [ 101 ]. |
| Bloqueadores de los receptores de angiotensina II tipo 1 (BRA) |
| Telmisartan |
Inhibe la proliferación celular y el crecimiento tumoral del carcinoma de células escamosas de esófago al inducir la detención del ciclo celular en la fase s [ 102 ]. |
| Candesartán |
Previene el crecimiento del cáncer de vejiga en un modelo de ratón al inhibir la angiogénesis, y el tratamiento combinado con candesartán y paclitaxel mejora la citotoxicidad inducida por paclitaxel [ 103 ].
El tratamiento con candesartán sensibiliza significativamente las células de adenocarcinoma de pulmón humano a la apoptosis mediada por ligando inductora de apoptosis relacionada con el factor de necrosis tumoral [ 104 ]. |
| Losartán |
Puede inhibir el crecimiento tumoral en el modelo de cáncer gástrico y suprimir la angiogénesis del tumor disminuyendo las expresiones de VEGF [ 99 ].
Puede ejercer actividad anti-metastásica inhibiendo la señalización del receptor de quimiocinas tipo 2 (CCR2) y suprimiendo el reclutamiento de monocitos en un modelo de ratón con tumores e indirectamente como efecto antiinflamatorio e independientemente de AT1R [ 105 ].
Mejora la angiogénesis, la inflamación y la inducción de estrés oxidativo a través del receptor de angiotensina II tipo 1 (AT1R) en un modelo murino de metástasis pulmonar de cáncer colorrectal [ 106 ].
Inhibe el crecimiento celular, disminuye la expresión de c-myc y aumenta la apoptosis en líneas celulares leucémicas [ 101 ]. |
| Valsartán |
Puede inhibir el crecimiento tumoral en el modelo de cáncer gástrico y suprimir la angiogénesis del tumor, disminuyendo las expresiones de VEGF [ 99 ]. |
| Antagonistas de la aldosterona |
| Espironolactona |
Inhibe el crecimiento de células cancerosas y es altamente tóxico para las células madre cancerosas; altera la reparación de roturas de doble hebra del ADN e induce la apoptosis en las células cancerosas y las células madre cancerosas (CSC), al tiempo que evita las células sanas. In vivo, este tratamiento reduce el tamaño y el contenido de CSC de los tumores [ 107 ]. |
This entry is adapted from the peer-reviewed paper 10.3390/ijms22137106