Heart failure (HF) and cancer are the main public health issues in industrialized countries and are increasing in prevalence, especially, in the ageing population. These two diseases were thought to be independent; however, new research has revealed that cancer and HF frequently coexist in the same patient. Furthermore, as cancer-specific mortality decreases and the surviving population gets older, the overlap between cardiac disease and cancer patients is growing. As a result, the discipline of cardio-oncology has primarily focused on the adverse effects of anti-cancer therapy. HF is one of the most serious consequences of cardiotoxic cancer therapy.
- heart failure
- myocardial infarction
- cancer
- renin-angiotensin-aldosterone system
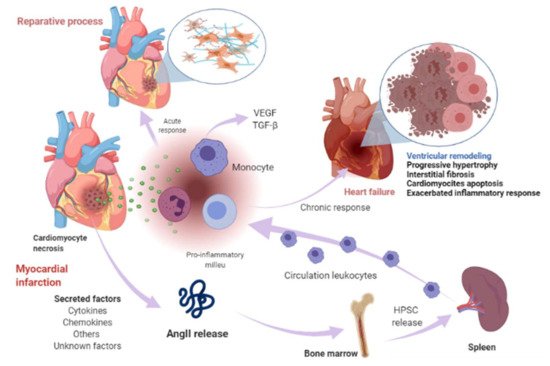
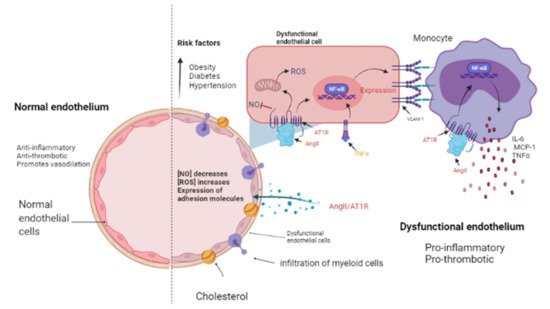
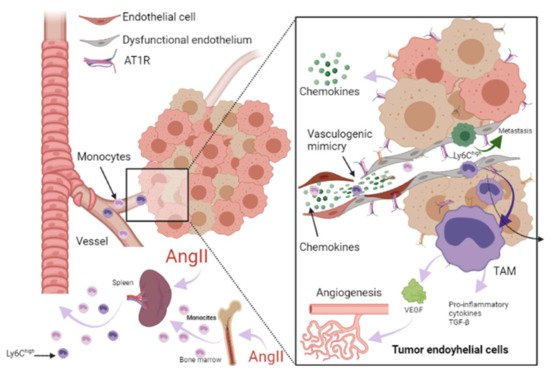
1. Renin-Angiotensin-Aldosterone System Linking Ischemic Heart Failure and Cancer
2. Papel de la farmacoterapia con inhibidores del RAAS en la insuficiencia cardíaca y el cáncer
| RAAS Inhibitor | Observations |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Long-term administration was associated with an improvement in survival and reduced morbidity and mortality due to major cardiovascular events in patients with asymptomatic left ventricular (LV) dysfunction after myocardial infarction (MI) [82]. |
| Enalapril | Increased exercise time and left ventricular ejection fraction (LVEF) [83]. |
| Perindopril | Increased 6 min walk distance but did not decrease mortality [84]. After 1-year treatment reduced progressive LV remodeling but it was not associated with better clinical outcomes [85]. |
| Ramipril | Administration to patients with clinical evidence of either transient or ongoing heart failure (HF) after MI resulted in a substantial reduction in premature death from all causes [86]. |
| Trandolapril | Long-term treatment in patients with reduced LV function soon after MI significantly reduced the risk of overall mortality, mortality from cardiovascular causes, sudden death, and the development of severe HF [87]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Telmisartan was well tolerated in patients unable to tolerate ACEI. Although the drug had no significant effect on hospitalizations for HF, it modestly reduced the risk of the composite outcome of cardiovascular death, MI, or stroke [88]. |
| Candesartan | Slightly decreased hospitalizations but did not decrease mortality [89]. Reduced cardiovascular mortality and hospital admissions for worsening chronic HF. Patients with reduced ejection fraction were the most benefited [90]. |
| Losartan | Reduced the rate of death or admission for HF in patients with HF, reduced LVEF, and intolerance to ACEI [91]. |
| Valsartan | In patients with MI associated with HF and/or LV dysfunction, valsartan administration in the immediate post MI period demonstrated equal efficacy than captopril [92,93]. |
| Aldosterone antagonists | |
| Spironolactone | Prevented LV fibrosis and remodeling after MI [94] |
| RAAS Inhibitor | Findings |
|---|---|
| Angiotensin converting enzyme inhibitors (ACEI) | |
| Captopril | Inhibits tumor growth in a gastric cancer model and suppresses the angiogenesis of the tumor by decreasing the expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP)-7 in a mouse model with human gastric cancer [99]. Attenuates cell migration in a breast cancer model [100]. Inhibits cell growth, decreases c-myc expression, and increases apoptosis on leukemic cell lines [101]. |
| Enalapril | Inhibits tumor progression and reduces number of tumor-associated macrophages (TAMs) [54]. |
| Perindopril | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor by decreasing the expression of VEGF and MMP-7 in a mouse model with human gastric cancer [99]. |
| Ramipril | Decreases systemic inflammation [55]. |
| Trandolapril | Inhibits cell growth, decreases c-myc expression, and increases apoptosis in leukemic cell lines [101]. |
| Angiotensin II type 1 receptor blockers (ARBs) | |
| Telmisartan | Inhibits cell proliferation and tumor growth of esophageal squamous cell carcinoma by inducing s-phase cell cycle arrest [102]. |
| Candesartan | Prevents bladder cancer growth in a mouse model by inhibiting angiogenesis, and combined treatment with candesartan and paclitaxel enhances paclitaxel-induced cytotoxicity [103]. Candesartan treatment significantly sensitizes human lung adenocarcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis [104]. |
| Losartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor decreasing the expressions of VEGF [99]. Can exert anti-metastatic activity by inhibiting chemokine receptor type 2 (CCR2) signaling and suppressing monocyte recruitment in a mouse model with tumors and indirectly as anti-inflammatory effect and independently of AT1R [105]. Ameliorates angiogenesis, inflammation and the induction of oxidative stress via type-1 angiotensin-II receptor (AT1R) in a murine model of lung metastasis of colorectal cancer [106]. Inhibits cell growth, decreases c-myc expression and increases apoptosis in leukemic cell lines [101]. |
| Valsartan | Can inhibit the tumor growth in gastric cancer model and suppress the angiogenesis of the tumor, decreasing the expressions of VEGF [99]. |
| Aldosterone antagonists | |
| Spironolactone | Inhibits cancerous cell growth and is highly toxic for cancer stem cells; impairs DNA-double-strand breaks repair and induces apoptosis in cancer cells and cancer stem cells (CSCs) while sparing healthy cells. In vivo, this treatment reduces the size and CSC content of tumors [107]. |
This entry is adapted from the peer-reviewed paper 10.3390/ijms22137106