 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alexandar Tzankov | + 303 word(s) | 303 | 2021-02-16 10:19:08 | | | |
| 2 | Conner Chen | + 780 word(s) | 1083 | 2021-02-22 11:21:39 | | | | |
| 3 | Conner Chen | Meta information modification | 1083 | 2021-02-23 10:00:03 | | |
Video Upload Options
Hodgkin lymphoma (HL) is composed of many reactive and only a few cancer cells, so-called Hodgkin and Reed-Sternberg (HRS) or lymphocyte predominant (LP) cells.
1. Searching and Finding the Needle in the Haystack
Hodgkin lymphoma (HL) is predominantly composed of reactive, non-neoplastic cells surrounding scarcely distributed tumor cells, that is, so-called Hodgkin and Reed-Sternberg (HRS) or lymphocyte predominant (LP) cells. This scarcity impeded the analysis of the tumor cell genomes for a long time, but recently developed methods (especially laser capture microdissection, flow cytometry/fluorescence-activated cell sorting) facilitated molecular investigation, elucidating the pathophysiological principles of “Hodgkin lymphomagenesis”. The molecular cornerstones of this Hodgkin lymphomagenesis can be summarized as follows (Figure 1): Firstly, the malignant cells of HL evade the immune system by altered expression of PDL1/2, B2M and MHC class I and II due to various genetic alterations. Secondly, tumor growth is promoted by permanently activated JAK/STAT signaling due to pervasive mutations of multiple genes involved in the pathway. Thirdly, apoptosis of neoplastic cells is prevented by alterations of NF-κB compounds and the PI3K/AKT/mTOR axis. Additionally, Epstein-Barr virus infection can simultaneously activate JAK/STAT and NF-κB, similarly leading to enhanced survival and evasion of apoptosis. Finally, epigenetic phenomena such as promoter hypermethylation lead to the downregulation of B-lineage-specific, tumor-suppressor and immune regulation genes.
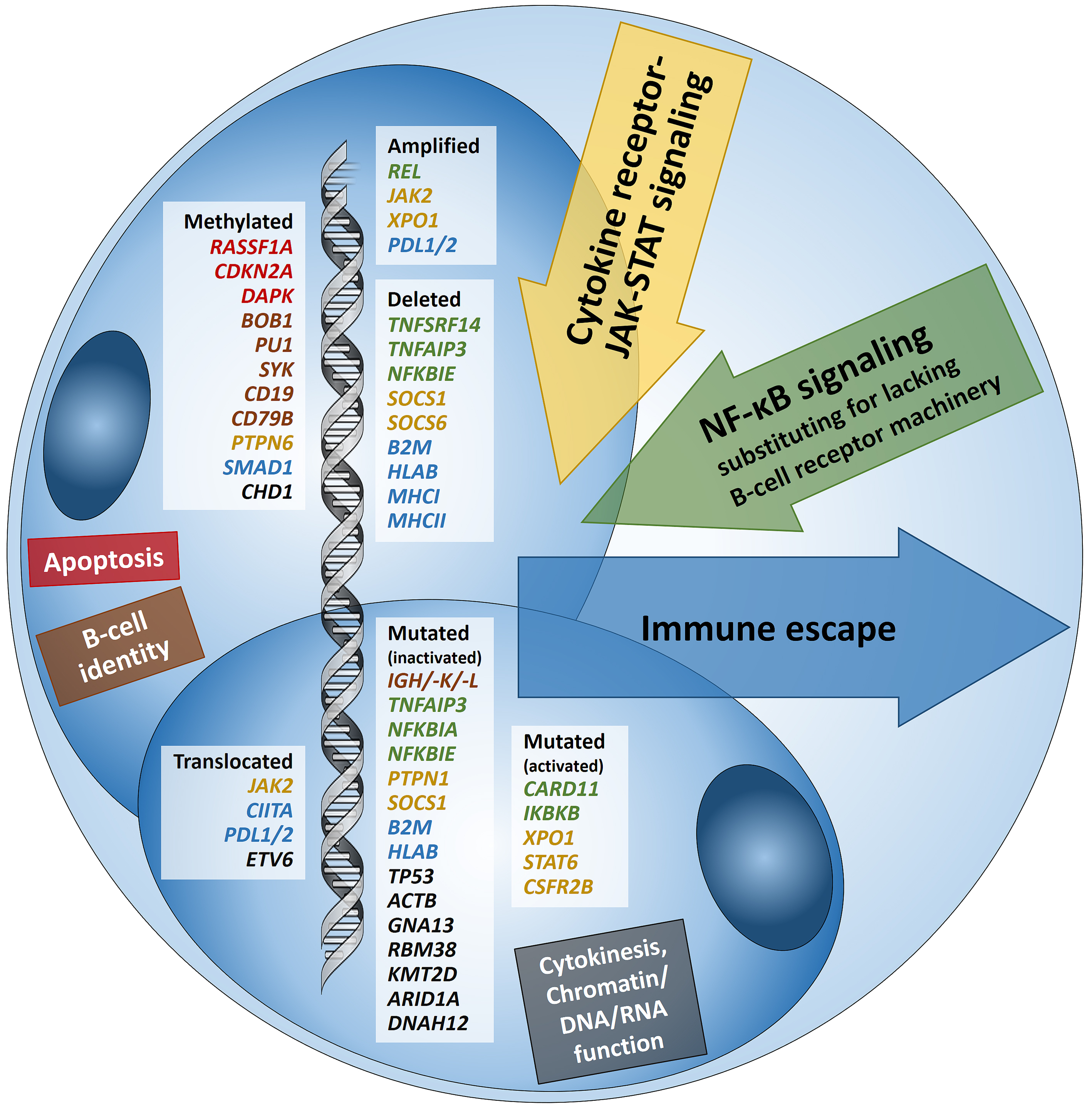
Figure 1: Summary of known genetic aberrations in classic Hodgkin lymphoma (cHL) arranged according to aberration type and color-coded according to the affected cellular process that they dysregulate in Hodgkin and Reed-Sternberg cells; genes encoding for proteins related to apoptosis are in red, to B-cell identity—in brown, to cytokine (mainly JAK-STAT) signaling—in orange-gold, to NF-κB signaling—in green, to immune escape—in blue, and to cytokinesis, chromatin/DNA/RNA functions—in black.
2. Immune Evasion
Several genes involved in the development of hematolymphoid malignancies are located at the 9p24 locus. This includes key targets of immune checkpoint inhibition such as programmed death ligands 1 and 2 (PDL1/PDL2), which evokes potential therapeutic interest (rev. in [1] and in other contributions within this special issue). Most investigated cases of cHL show genetic alterations of PDL1/2, most commonly copy number gains and amplifications (up to 55% and 35%, respectively) [2]. In cHL—mainly the nodular sclerosis subtype—these copy number gains were found to correlate with higher expression of PDL1 as determined by immunohistochemistry (Figure 2A). They represent the hallmark of tumor-induced immune modulation mainly impeding effector T-cell proliferation and activation as well as stimulating immunosuppressive regulatory T-cells [1][2][3][4]. In a minority of cases, structural abnormalities of 9p24 have been found in cHL (Figure 2B, insert), leading to translocations of PDL1/2 to several partners [5]. Single nucleotide mutations, insertions and deletions of PDL1/2 are not (yet) found to play a major role in lymphomagenesis [1].
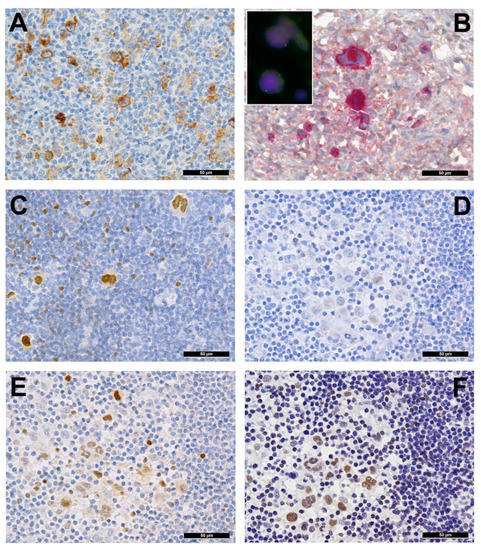
Figure 2. (A) PDL1 overexpressing Hodgkin and Reed-Sternberg (HRS) cells in a case of PDL1/2 amplified classic Hodgkin lymphoma (cHL). (B) HRS cells expressing phosphorylated (p) JAK2 in a JAK2 rearranged cHL (insert with split red and green FISH signals corresponding to the rearranged allele and one fused yellow signal corresponding to the wild type allele of the JAK2 gene in the respective large HRS cell-equivalents utilizing a break-apart JAK2 probe). (C) pSTAT6 overexpressing HRS cells in a case of STAT6-mutated cHL. (D) Expression of pSTAT6 only in a few HRS cells as compared to. (E) pSTAT5 and, particularly, pSTAT3. (F) in a case of SOCS1-mutated cHL.
Importantly, 9p24 alterations, especially copy number gains, were associated with inferior outcome in conventionally treated patients [2] but were an indicator for response and superior progression-free survival—to PD1/PDL1 immune checkpoint inhibition-based immunotherapy [6]. Along with very convincing results of a prospective trial with nivolumab in relapsed or refractory cHL [7], this was founding for treatment advances in such instances as nicely reviewed [8], and may lead to first-line therapy paradigm changes in cHL. Furthermore, a very recent work highlighted the importance of a broad baseline T-cell repertoire for successful immune-checkpoint inhibitor treatment, being most effective in patients with therapy-associated diversity increase in the CD4+ compartment and in those with an abundance of activated natural killer cells and a newly identified CD3−CD68+CD4+GrB+ subset of innate immune cells, which may function as direct cytotoxic effectors in even the absence of major histocompatibility complex (MHC) class I (the latter being characteristic of cHL; see below) [9].
Inactivating mutations of the beta 2 microglobulin gene (B2M) also play an instrumental role in immune evasion, influencing the assembly of MHC class I and thus altering tumor cell “visibility” for effector cells [1]. Indeed, B2M is the most commonly mutated or deleted gene in up to 70% of studied cHL cases [10][11]. Furthermore, its deficiency is associated with the nodular sclerosis subtype, pointing towards its potential influence on the tumor microenvironment [7][9][12]. Wienand et al. additionally detected mutations or deletions of HLA-B in approximately 15% of cHL, representing another potential mechanism of MHC class I assembly dysregulation. Moreover, the MHCI (and MHCII) loci at 6p21 are among the commonly deleted in cHL [13][14][11]. Interestingly, a decrease of MHC class I expression is associated with inferior clinical outcome after standard chemotherapy, but not immune-checkpoint inhibition. To be comprehensive, Epstein-Barr virus (EBV) positive cHL have significantly higher MHC class I expression on HRS cells than EBV-negative cases [11][15]. In contrast to MHC class I, the expression of MHC class II is predictive for response to PD1-blockade in cHL [16], fitting well with the above-mentioned observations on the central role of immune responses linked to the CD4+ cellular compartment.
Finally, the MHC class II transactivator CIITA has been identified to be involved in a gene fusion in cHL cell lines and in 15% of investigated clinical cases [17]. Genomic aberrations in CIITA result in a downregulation of surface MHC class II expression as well as overexpression of PDL1/PDL2, hampering anti-tumor immune response.
A complex network of cytokines and chemokines secreted by both malignant and reactive cells orchestrates the interaction between HRS and LP cells, respectively, and the surrounding microenvironment [12]. One component of this network is the immunosuppressive effect of transforming growth factor-beta (TGF-β) on tumor-infiltrating lymphocytes. Until recently, it was unclear why HRS and LP cells remain unaffected by the anti-neoplastic properties of TGF-β. Previous studies on diffuse large B-cell lymphoma (DLBCL) revealed SMAD1 as a key messenger in the tumor-suppressive signaling axis of TGF-β [18]. In concordance with this study, our group was able to show a lack of SMAD1 expression due to hypermethylation of its promoter region in LP and HRS cells of almost all studied clinical cases (14/14 NLPHL cases, 100% and 138/143 cHL cases, 97%). Most interestingly, this mechanism was reversible in an affected cell line by treatment with decitabine, a DNA methyltransferase inhibitor [19].
References
- Menter, T.; Tzankov, A. Genetic alterations of 9p24 in lymphomas and their impact for cancer (immuno-)therapy. Virchows Arch. 2019, 474, 497–509, doi:10.1007/s00428-018-2438-6.
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697, doi:10.1200/JCO.2016.66.4482.
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029, doi:10.1084/jem.20090847.
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induc-tion via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277, doi:10.1182/blood-2010-05-282780.
- van Roosbroeck, K.; Ferreiro, J.F.; Tousseyn, T.; van der Krogt, J.A.; Michaux, L.; Pienkowska-Grela, B.; Theate, I.; De Paepe, P.; Dierickx, D.; Doyen, C.; et al. Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid ma-lignancies. Genes Chromosomes Cancer 2016, 55, 428–441, doi:10.1002/gcc.22345.
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950, doi:10.1200/JCO.2017.77.3994.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319, doi:10.1056/NEJMoa1411087.
- Voorhees, T.J.; Beaven, A.W. Therapeutic updates for relapsed and refractory classical Hodgkin lymphoma. Cancers (Basel) 2020, 12, 2887, doi:10.3390/cancers12102887.
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.H.; Weirather, J.L.; et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020, 26, 1468–1479, doi:10.1038/s41591-020-1006-1.
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072, doi:10.1182/blood-2014-11-610436.
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019, 3, 4065–4080, doi:10.1182/bloodadvances.2019001012.
- Nagpal, P.; Descalzi-Montoya, D.B.; Lodhi, N. The circuitry of the tumor microenvironment in adult and pediatric Hodgkin lymphoma: Cellular composition, cytokine profile, EBV, and exosomes. Cancer Rep. (Hoboken) 2020, ahead of print, doi:10.1002/cnr2.1311.
- Hartmann, S.; Martin-Subero, J.I.; Gesk, S.; Hüsken, J.; Giefing, M.; Nagel, I.; Riemke, J.; Chott, A.; Klapper, W.; Parrens, M.; et al. Detection of genomic imbalances in microdissected Hodgkin and Reed-Sternberg cells of classical Hodgkin’s lym-phoma by array-based comparative genomic hybridization. Haematologica 2008, 93, 1318–1326, doi:10.3324/haematol.12875.
- Juskevicius, D.; Jucker, D.; Dietsche, T.; Perrina, V.; Rufle, A.; Ruiz, C.; Dirnhofer, S.; Tzankov, A. Novel cell enrichment technique for robust genetic analysis of archival classical Hodgkin lymphoma tissues. Lab. Investig. 2018, 98, 1487–1499, doi:10.1038/s41374-018-0096-6.
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916, doi:10.1158/2326-6066.
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950, doi:10.1200/JCO.2017.77.3994.
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381, doi:10.1038/nature09754.
- Stelling, A.; Wu, C.T.; Bertram, K.; Hashwah, H.; Theocharides, A.; Manz, M.G.; Tzankov, A.; Müller, A. Pharmacological DNA demethylation restores SMAD1 expression and tumor suppressive signaling in diffuse large B-cell lymphoma. Blood Adv. 2019, 3, 3020–3032, doi:10.1182/bloodadvances.2019000210.
- Gerlach, M.M.; Stelling-Germani, A.; Wu, C.T.; Newrzela, S.; Döring, C.; Vela, V.; Müller, A.; Hartmann, S.; Tzankov, A. SMAD1 promoter hypermethylation and lack of SMAD1 expression in Hodgkin lymphoma: A potential target for hypo-methylating drug therapy. Haematologica 2021, 106, 619–621, doi:10.3324/haematol.2020.249276.
- Menter, T.; Tzankov, A. Genetic alterations of 9p24 in lymphomas and their impact for cancer (immuno-)therapy. Virchows Arch. 2019, 474, 497–509, doi:10.1007/s00428-018-2438-6.
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029, doi:10.1084/jem.20090847.
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induc-tion via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277, doi:10.1182/blood-2010-05-282780.
- van Roosbroeck, K.; Ferreiro, J.F.; Tousseyn, T.; van der Krogt, J.A.; Michaux, L.; Pienkowska-Grela, B.; Theate, I.; De Paepe, P.; Dierickx, D.; Doyen, C.; et al. Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid ma-lignancies. Genes Chromosomes Cancer 2016, 55, 428–441, doi:10.1002/gcc.22345.
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950, doi:10.1200/JCO.2017.77.3994.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319, doi:10.1056/NEJMoa1411087.
- Voorhees, T.J.; Beaven, A.W. Therapeutic updates for relapsed and refractory classical Hodgkin lymphoma. Cancers (Basel) 2020, 12, 2887, doi:10.3390/cancers12102887.
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.H.; Weirather, J.L.; et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020, 26, 1468–1479, doi:10.1038/s41591-020-1006-1.
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916, doi:10.1158/2326-6066.
- Nagpal, P.; Descalzi-Montoya, D.B.; Lodhi, N. The circuitry of the tumor microenvironment in adult and pediatric Hodgkin lymphoma: Cellular composition, cytokine profile, EBV, and exosomes. Cancer Rep. (Hoboken) 2020, ahead of print, doi:10.1002/cnr2.1311.
- Stelling, A.; Wu, C.T.; Bertram, K.; Hashwah, H.; Theocharides, A.; Manz, M.G.; Tzankov, A.; Müller, A. Pharmacological DNA demethylation restores SMAD1 expression and tumor suppressive signaling in diffuse large B-cell lymphoma. Blood Adv. 2019, 3, 3020–3032, doi:10.1182/bloodadvances.2019000210.

