 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shih-Min Hsia | + 1997 word(s) | 1997 | 2021-01-18 10:10:21 | | | |
| 2 | Lily Guo | + 2760 word(s) | 4757 | 2021-02-08 06:48:54 | | |
Video Upload Options
Isoliquiritigenin (ISL), a natural bioactive compound with a chalcone structure, demonstrates high antitumor efficacy.
1. Indroduction
ISL is a flavonoid with a simple chalcone structure. The structure of ISL and its metabolites are shown in Figure 1. The previous studies demonstrated the six metabolites detected in phase I[1][2][3], including liquritigenin (M1), 2′,4,4′,5′-tetrahydroxychalcone (M2), sulfuretin (M3), butein (M4), davidigenin (M5), and cis-6,4′-dihydroxyaurone (M6). Among the six metabolites, butein is the more active metabolite in the liver and in HT22 cells, with significant distribution on M1, M3, and M4 (Figure 1) [1][2][4]. Moreover, the previous study reported that the dominant metabolites of ISL are THC (2,4,2′,4′-tetrahydroxychalcone) and naringenin chalcone in lung cells [5] In vivo absorption of ISL occurs in the intestines, transported to the liver for phase II biotransformation[2]. In phase II metabolism, liquiritigenin, glucuronidated ISL, glucuronidated liquiritigenin, and glucuronidated ISL are produced. Only glucuronidated liquiritigenin is predominant[6]. Many studies have suggested that secondary metabolites are involved in different biological activities and pharmaceuticals[1][2][6][7]. Therefore, these metabolites may differ in various cell lines or organs; however, they all share a similar structure to that of chalcone, which contains two aromatic rings connected by an unsaturated carbon chain, resulting in interconnected biological activities.
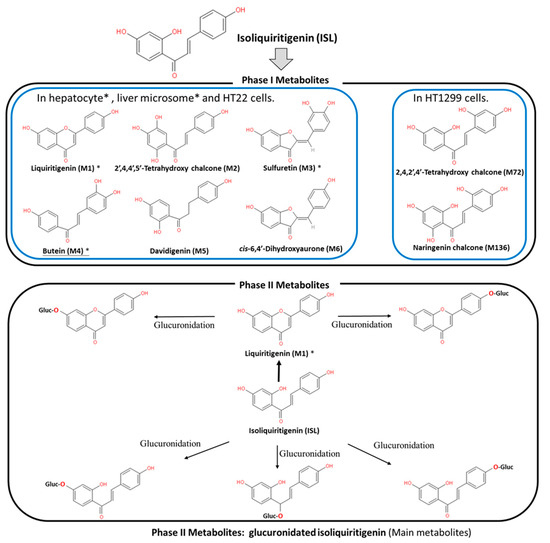
Figure 1. Metabolites of isoliquiritigenin (ISL). Phase I ISL metabolites were identified to be liquiritigenin (M1), 2′,4,4′,5′-tetrahydroxychalcone (M2), sulfuretin (M3), butein (M4), davidigenin (M5), and cis-6,4′-dihydroxyaurone (M6). Phase II metabolites were glucuronide conjugated process. Note: Figure was modified from[1][2].
2. ISL Pharmacokinetics
Evaluation of the safety of ISL is necessary for future clinical applications. Therefore, many studies, through different routes of administrations, including intravenously (IV), via hypodermic (IH) or intraperitoneal (IP) injection, and orally, have indicated that ISL exhibits a robust absorption capacity (absorption rate: ~60–90 min; oral absorption: >90%) with a strong elimination ability (t1/2: 2–4.9 h)[6][8][9][10]. Moreover, the data showed similar trends among different analytic methods, including high-performance liquid chromatography (HPLC), HPLC–MS/MS, and fluorescence spectrometry (SFS)[6][8][9]. This means that the absorption of ISL is quickly and widely distributed throughout the body[6][8][9][10]. Concentrations of ISL may vary in different tissues, including the heart, liver, lungs, spleen, kidneys, brain, muscles, and fat. ISL distribution mainly relies on the blood circulation, with the brain showing the lowest level of ISL due to the blood–brain barrier (BBB). These results imply that ISL is able to penetrate the BBB and exhibits neuroprotective activity in a male middle cerebral artery occlusion (MCAO)-induced focal cerebral ischemia rat model and high fat diet (HFD)-induced ICR mice model[11][12]. Interestingly, only after oral administration does [ISL]plasma exhibit a double-peak of ISL[10][13][14][15], the possible mechanism for which has been proposed as enterohepatic recycling. As a matter of fact, oral administration has become the most advanced application route.
3. ISL Nanoformulations and ISL Derivatives: Improved Efficacy
Generally speaking, poor bioavailability, rapid degradation, fast metabolism, and systemic elimination are the essential factors that lead to insufficient bioavailability. Insufficient bioavailability of ISL means that its efficacy is far less than 20%[6] [10]. The term insufficient bioavailability implies that patients show intolerance to bulk administration of ISL to reach the desired effect, thereby highlighting the need to improve its effectiveness. To improve solubility, enhancing its bioavailability and distribution, encapsulated ISL nanoparticles or nano-ISL have been developed. Below, we summarize various ISL nanoparticles applied in preclinical studies, for example, polymer nanoparticles, liposomes, micelles, solid lipid nanoparticles (SLNs), and polymer conjugates.
-
Nanosuspension: ISL is milled with HPC (hydroxypropyl cellulose) SSL and PVP (polyvinylpyrrolidone) K30 to form a lamelliform or ellipse shape of the nanosuspension. HPC SSL and PVP K30 act as stabilizer. These two nanosuspension particles (size: 238.1 ± 4.9 nm with SSL; 354.1 ± 9.1 nm with K30) do not only improve the solubility issue, but also enhance the cytotoxicity a 7.5–10-fold[16].
-
Nanoencapsulation: Mesoporous silica nanoparticles (MSNs) are a solid material, acting as a biodegradable nanoscale drug carrier. When MSNs are encapsulated with ISL, they improve the efficacy of ISL in vitro and in vivo[17].
-
Lipid–polymer hybrid nanoparticle system:
- 3.1.
-
iRGD hybrid NPs: The composition of lipid–polymer hybrid nanoparticles (NPs) include lactic-co-glycolic acid (PLGA), lecithin, and a hydrophilic poly-ethylene-glycol (PEG). ISL-loaded hybrid NPs are composed of an inner PLGA core with an outer lipid layer (PEG, lecithin, and iRGD peptides). iRGD peptides (CRGDK/RGPD/EC, a tumor-homing peptides), can deliver drugs to a tumor. In vitro, ISL–iRGD NPs show stronger inhibition effects and induce apoptosis effects. In vivo, ISL–iRGD NPs show stronger effects in the viability of tumor cells. Herein, iRGD-modified lipid–polymer NPs showed better solubility, bioavailability, and targeting distribution[18].
- 3.2.
-
Hydrophilic polyanion solid lipid nanoparticles (SLNs): SLNs are composed of natural lipids such as lecithin or triglycerides that remain solid at 37 °C. SLNs can protect labile compounds from chemical degradation and can improve bioavailability. Low-molecular-weight heparins (LMWHs) are fragments of heparin showing hydrophilic polyanions that can improve the efficacy of ISL[19].
-
Microemulsion: The self-microemulsifying drug delivery system (SEMDDS) was designed for improving the solubility, absorption, and bioavailability of lipophilic drugs. The SMEDDS comprises ethyl oleate (EO; oil phase), Tween 80 (surfactant), and PEG 400 (co-surfactant). ISL-loaded SMEDDS has been proven to improve the solubility and oral in vivo availability[13].
-
ISL-loaded nanostructured lipid carriers (ISL-NLCs): NLCs mix solid lipids with spatially incompatible liquid lipids, which leads to a special nanostructure with improved properties for drug loading. ISL-loaded NLCs are constructed by glycerol monostearate (MS) and Mi-glyol-812 as the solid and liquid lipid materials to carry the ISL[20]. In pharmacokinetic studies, less than 10% of the NLCs remains in the stomach after oral administration, mainly absorbed in the colon[19]. Moreover, the antitumor effect of ISL-loaded NLCs has been evaluated in sarcoma 180 (S180)-bearing and murine hepatoma (H22)-bearing mice models via IP administration[20]. A biodistribution study showed that the ISL concentration of ISL-loaded NLCs in the tumor is higher 2.5-fold than free ISL. In a skin permeability study, the previous study suggested NLCs as a promising carrier to deliver the ISL[21].
-
TPGS-modified proliposomes: D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) has been selected as an excipient for ISL-loaded TPGS-modified proliposomes (ISL-TPGS-PLP), prepared using the film dispersion method with ISL-loaded proliposomes (ISL–PLP). ISL-TPGS-PLP can enhance the solubility, bioavailability and liver-targeting ability of ISL[14].
-
Polymeric micelles: PEO (polyethylene oxide)–PPO (polypropylene oxide)–PEO (polyethylene oxide) triblock copolymers are highly biocompatible and act as surface-active agents. P123 (PEO20–PPO65–PEO20) can remarkably enhance the retention of poorly soluble drugs in the blood circulation. Another important derivative of Pluronic, F127 (PEO100–PPO69–PEO100), possesses high biocompatibility. Therefore, mixed F127/P123 polymeric micelles have been developed, which have remarkably enhanced bioavailability with high encapsulation efficiency and low particle size. ISL-loaded F127/P123 polymeric micelles (ISL-FPM) improve the solubility as well as enhance the bioavailability and antioxidant activity of ISL [22].
-
Nanoliposomes (NLs): Drug-loaded PEGylated nanomaterials have shown effective cancer cell-killing ability, PEG2000-DPSE-QUE-NLs (polyethyleneglycol-2000-distearoyl phosphatidyl ethanolamine loaded with querce-tin (QUE)) can efficiently disperse in aqueous media compared to controls, and PEGylated (PEG2000-DPSE) NLs have been found to be effective drug delivery vehicles when simply loaded with ISL. ISL-NLs as tumor-targeted drug carriers are more effective in regulating glycolysis in colon cancer cell lines (CRC: HCT116)[23].
-
Hydrogel: Hydrogels are composed of hyaluronic acid (HA) and hydroxyethyl cellulose (HEC), and they can improve the skin permeation of ISL[24].
As described above, many experiments have been conducted to evaluate the various properties of ISL nanoformulation have been developed to address the problems of bioavailability and solubility. Nanoformulation studies have been conducted in vitro and in vivo (Table 1), demonstrating that ISL nanoformulations improve the bioavailability by 2–10-fold[13][20][22].
Table 1. Nano-formulation of ISL.
| Formulation | Material | Particle Size (nm) |
Model | Conclusion | Ref |
|---|---|---|---|---|---|
| Nanosuspension | Hydroxypropyl cellulose-SSL Polyvinylpyrroli-done-K30 |
238.1 ± 4.9 354.1 ± 9.1 |
In vitro: A549 | HPC SSL‑ISL‑NS and PVP K30-ISL‑NS both improve the solubility and cytotoxic activity of ISL (IC50: ~0.08 µM). | [16] |
| Nanoencapsulation | Mesoporous silica nanoparticles | ~200 | In vitro: mouse primary bone marrow-derived macrophages (BMMs) In vivo: lipopolysaccharide (LPS)-mediated calvarial bone erosion model (received 50 mg/kg MSNs-ISL; once every 2 days via subcutaneous injection) Experiment period: 7 days |
MSNs-ISL as an effective natural product-based bone-bioresponsive nanoencapsulation system prevents osteoclast-mediated bone loss (In vitro effective dose: 16~64 µg/mL). | [17] |
| Lipid–polymer hybrid | ISL-iRGD nanoparticles | ~130 138.97 ± 2.44 |
In vitro: MCF-7, MDA-MB231, 4T1 In vivo: 4T1-bearing nude mouse (received 35 µg/kg once every 2 days via IV injection) Experiment period: 20 days |
RGD modified lipid–polymer hybrid NPs improve ISL in anti-breast cancer efficacy (Effective dose: >12 µM). | [18] |
| LMWH-ISL-SLN | 217.53 ± 4.86 | In vitro: HepG2 In vivo: Kunming mice (6 female and 6 male; 50 mg/kg via IV injection daily) Experiment period: 14 days |
Pharmacokinetics of LMWH-ISL-SLN demonstrated its safety and better bio-distribution after intravenous administration (In vitro IC50: ~7.45 µg/mL). | [19] | |
| Micro-emulsion | Self-microemulsifying drug delivery system (SEMDDS) | 44.78 ± 0.35 | In vivo: SD rat (oral administration: a single dose: 200 mg/kg) Experiment period: 24 h |
ISL-SMEDDS can enhance the solubility and oral bioavailability of ISL. |
[13] |
| 20.63 ± 1.95 | In vivo: SD rat (oral administration: twice a day; 20 mg/kg) Experiment period: 63 days |
[25] | |||
| Nanostructured lipid carrier (ISL-NLC) | Monostearate and lecithin | 160.73 ± 6.08 | In vivo: Kunming mice bearing H22 and S180 tumor (intraperitoneal injection daily) Experiment period: 12 days | ISL-NLC nanoparticles with high envelopment efficiency with initial burst release, exhibiting superior in vivo antitumor effect and biodistribution. | [20] |
| MS and Miglyol 812 | 160.73 ± 6.08 | In vivo: SD rat (oral administration: a single dose: 20 mg/kg) Experiment period: 36 h |
NLC are valuable as an oral delivery carrier to enhance the absorption of a poorly water-soluble drug, ISL. | [15] | |
| Ceramide, cholesterol, caprylic/capric triglyceride | 150.2–251.7 | In vitro: Franz diffusion cell In vivo: ICR mice |
NCL improved the skin permeation of ISL (permeability: 8.48~10.12 μg/cm3). |
[21] | |
| TPGS-modified proliposomes | D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), proliposomes |
23.8 ± 0.9 | In vivo: Swiss-ICR mice oral administration Experiment period: 24 h |
ISL-TPGS-PLP had small particle size, high encapsulation efficiency and drug loading capacity, and possessed good storage stability. | [14] |
| Polymeric micelles | ISL-loaded F127/P123 polymeric micelles (ISL-FPM) | 20.12 ± 0.72 | In vivo: SD rat, (oral administration: a single dose 200 mg/kg) Experiment period: 24 h |
ISL-FPM act as a promising approach to improve solubility as well as enhance bioavailability and antioxidant activity of ISL. | [22] |
| Liposome | Phospholipid and cholesterol | 233.1 | In vitro: HeLa and SiHa | ISL liposome can significantly inhibit the proliferation of human cervical cancer cells in vitro. | [26] |
| Nanoliposome | Sodium cholate, cholesterol and IPM were melted with a ratio of 5:1:4 (w/w/w) | 82.3 ± 35.6 | In vitro: HCT116 and HT29 | ISL involved in the glucose metabolism in colon cancer. | [23] |
| Hydrogel systems | HA-HEC hydrogels | N.A. | In vitro: skin permeation study Franz diffusion cells | HA-HEC hydrogel showing the stable viscoelastic be haviour and the optimal adhesiveness has potential to enhance skin permeation of IS (permeability: 20 μg/cm3). | [24] |
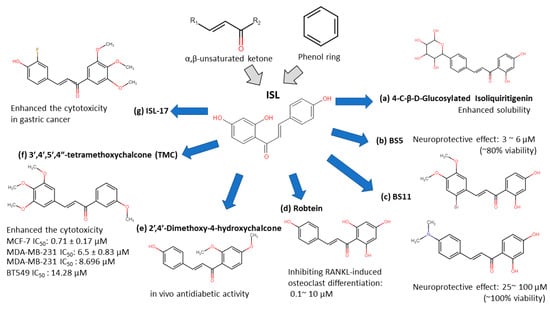
Figure 2. Isoliquiritigenin (ISL) derivatives.
-
4-C-β-D-glucosylated ISL (Figure 2a): Glucosylation of low molecular weight compounds have improve water solubility and bioavailability with a good inhibition of aldose reductase (AR)[33].
-
Synthetic isoliquiritigenin derivatives (BS5 and BS11 in Figure 2b,c): The compounds BS5 and BS11 with m-, p-dimethoxy, o-bromo phenyl group shows neuroprotective effects at 3 μM to 6 μM with higher viability (~80–100%)[32].
-
Robtein (ISL-derivative #10; Figure 2d): Robtein exhibited osteoclast differentiation and activation without any significant changes of viability or cytotoxicity[28].
-
2′,4′-dimethoxy-4-hydroxychalcone (Figure 2e): shows in vivo antidiabetic activity[31].
-
3′,4′,5′,4″-tetramethoxychalcone (TMC; Figure 2f): Introducing methylation of hydroxy groups significant increase cytotoxic activity in breast cancer[27], especially targeting on triple-negative breast cancer (TNBC)[29].
-
ISL-17 (Figure 2g): A fluorine atom was introduced to the structure of ISL named ISL-17 showed the anti-tumor activities in gastric cancer[28].
However, the poor bioavailability and water-solubility issues remain in clinical applications. Future studies are still needed to elucidate the ISL formulations that would be more suitable for human clinical trials.
4. ISL Docking Model
ISL had been reported to exert diverse biological properties, but the specific molecular interaction that underlies these activities has not been fully unveiled. Based on molecular docking analysis, many studies have proposed that ISL has a direct interaction in different molecules (Figure 3), such as SIRT1 [34]VEGF2 receptor[35], GRP78[36], FLT3[37], EGFR[38], IKKβ[39], Toll-like receptors (TLRs)[40], CK-2 (IC50: 17.3 µM)[41], H2R[42], COX-2[43], aromatase (Ki: 2.8 µM)[44][45], topoisomerase I [46] and DNMT1[47]. These docking results imply that the binding pocket is composed of hydrophobic regions and is stabilized by a hydrogen bond with its neighboring carbonyl group. The hydrogen bond interactions and π–π stacking contribute to a tight interaction with the binding site. These docking results provide valuable information about the binding interactions of ISL and the active site, although more studies are required to approve them. Using a bioassay-guided purification method, suggested that isolated ISL acts as a xanthine oxidase inhibitor (IC50: 55.8 µM; Ki: 17.4 µM) to avoid transplantation rejection and ischemia reperfusion damage [48]. In brief, multiple docking candidates indicate that ISL exhibits multiple biological properties and serves as a potential lead compound for developing new therapy in cancer treatment.
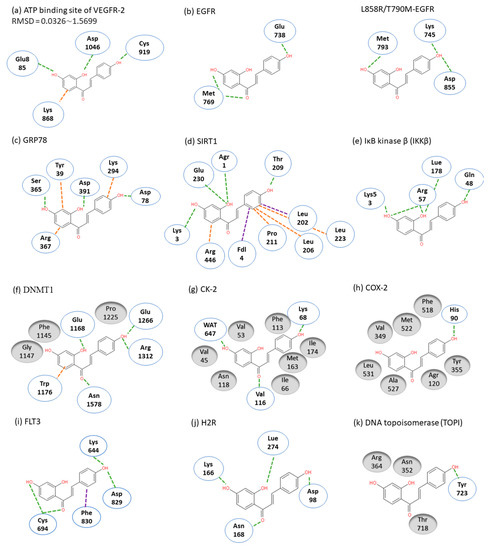
Figure 3. Molecular docking models. Interactions are represented in green (hydrogen bonding), orange (π–π stacking), purple (sigma-π) dash lines and gray (hydrophobic interaction: Van der Waals). (a) VEGFR-2; (b) EGFR; (c) GRP78; (d) SIRT1; (e) IKKβ; (f) DMNT1; (g) CK-2; (h) COX-2; (i) FLT3; (j) H2R; (k) TOPI.
5. ISL Biology Effects
In targeting cancers, ISL possesses various biologic activities, such as anti-inflammation, antioxidation, antiviral, antidiabetic, neuroprotective effect, chemopreventive, and antitumor growth properties (Figure 4 and Figure 5). A selective cytotoxicity effect of ISL has been reported (Table 2 and Table 3), and the effective dose in tumor cell lines shows very little cytotoxic effect on normal cells. Most studies have claimed that ISL significantly inhibits the viability of cancer cell but has little toxicity on normal cells. For example, Wu et al. (2017) compared the human endometrial stromal cells (T-HESCs; as a control) and human endometrial cancer cell lines (Ishikawa, HEC-1A, and RL95-2 cells). Their results indicated that ISL inhibits the growth of cancer cells at concentrations below 27 μM, but has little effect on normal cells[49]. Na et al. (2018) claimed that ISL shows little toxicity on normal hepatocyte cell lines (AML-12); only when applied in concentrations of over 100 μM is ISL harmful to normal hepatocytes [50]. Most studies have focused on the cytotoxicity between tumor and normal cells, and the effects of ISL on normal cells remain unknown. As Peng et al. (2015) mentioned, further research on the target organ toxicity or side effects of ISL is needed. The safety of ISL is always one of the most important concerns that must be evaluated.
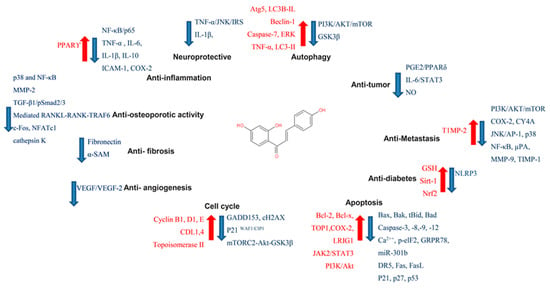
Figure 4. Pharmacological effect of ISL. The scheme presents the biological effects of ISL and molecular mechanisms of ISL against cancer via various signal pathways.
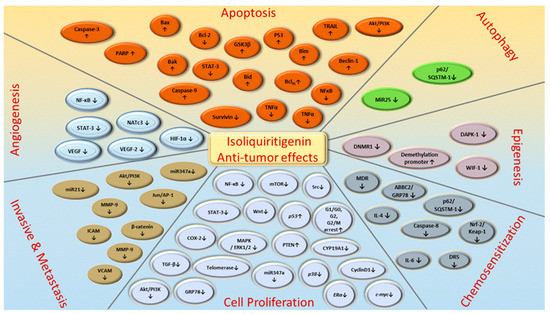
Figure 5. ISL-mediated regulation of molecular targets underlying anti-tumor effects, including tumor proliferation suppression, apoptosis induction, EMT/metastasis, epigenetic responses and sensitization to chemotherapy. Downward arrows (↓) represent downregulation while upward arrows (↑) represent upregulation. This figure was modified from[51].
Table 2. ISL influenced on normal cell lines.
| Type | Cell Line | Result | Ref |
|---|---|---|---|
| Breast | MCF-10A (0~50 µM) (24 h) |
ISL had no significant influence on MCF-10A as human normal tissues. | [36] |
| MCF-10A (0~100 µM) (24 h) |
ISL had limited inhibitory effects on the proliferation in normal cell and did not show the chemosensitization effect with epirubicin. | [52] | |
| H184B5F5/M10 (0.1~10 µM) (6~48 h) |
ISL did not influence the normal cell viability at the at 0.1~10 µM. | [53] | |
| Lung | HELF (24~72 h) |
Both pure drug of ISL and nanosuspension showed low toxicity to normal cells. | [16] |
| Hepatocyte | AML-12 (0~200 µM) (24 h) |
5~50 μM of ISL increased cell proliferation, strong cytotoxicity was observed over 100 μM. | [50] |
| Uterus Endometrium |
T-HESCs (5~100 µM) (24~48 h) |
The viability of T-HESCs showed significant changes when ISL concentration over 75 μM was applied. | [49] |
| Gastric | GES-1 (20 µM) (48 h) |
ISL exhibited a negligible effect on cell growth and cell viability exceeded 70%. | [28] |
| Endothelia | HUVEC | Over 10 µM of ISL is nontoxic with inhibiting the VCAM-1 and E-selectin. | [54] |
| Small intestine | IEC-6 (10~100 µM) (24 h) |
No effect was observed in IEC-6 cells. | [55] |
| Oral | SG cell (25~400 μM) (24 h) |
The half maximal effective dose (IC50) of ISL is 386.3 ± 29.7 μM. | [56] |
| Brain | H22 | ISL had the potential to against glutamate-induced neuronal cell death (neuroprotective effect) | [32] |
able 3. Different pathways of various cancers regulated by ISL.
| Type of Cancer | Cell | Testing Range/IC50 | Signaling Pathways Effect of ISL (In Vitro) |
Ref |
|---|---|---|---|---|
| Breast cancer | MCF-7 | Testing conc: 10 nM~10 µM (5 days; 10 nM is sufficient) |
|
[57] |
| MCF-7 MDA-MB-231 |
Effective conc: 25 µM and 50 µM (24 h) |
|
[41] | |
| MCF-7 MDA-MB-231 HUVEC |
Testing conc.: 0, 20, 40, 60, 80, 100 µM |
|
[58] | |
| Tumor cell line: MCF-7 IC50 estimated = ~33.39 µM MDA-MB-231 IC50 estimated = ~35.64 µM (48 h) |
||||
| HUVEC IC50 estimated = ~75.48 µM | ||||
| PMA-induced COX-2 in MCF-10A |
Effective conc: 0.1 µM and 10 µM (24 h; 1 µM is sufficient.) |
|
[59] | |
| BT549 MDA-MB-231 |
Effective conc.: 10, 20, 40 µM (12 h) |
|
[60] | |
| MDA-MB-231 Hs-578T |
Effective conc.: ~20 µM |
|
[61] | |
| Breast cancer | MCF-7 MDA-MB-231 |
Testing conc.: 0, 5, 10, 20 µM |
|
[62] |
| Tumor cell line: MCF-7 IC50 = 10.08 µM MDA-MB-231 IC50 = 5.5 µM (48 h) |
||||
| MCF-7 MDA-MB-231 |
Testing conc.: 0, 6.25, 12.5, 25, 50, 100 µM |
|
[63] | |
| Tumor cell line: MCF-7 IC50: 32.66 µM MDA-MB-231 IC50: 22.36 µM (24 h) |
||||
| MDA-MB-231 Hs-578T |
Effective conc.: 10 µM and 20 µM |
|
[64] | |
| MCF-7 MDA-MB-231 BT549 MCF-10 |
Testing conc.: 1, 5, 10 and 25 µM |
|
[36] | |
| Tumor cell lines: MCF-7 IC50 estimated: ~33.0 µM MDA-MB-231 IC50 estimated: ~21.2 µM BT549 IC50 estimated: ~18.1 µM (24 h) |
||||
| Normal cell line: MCF- 10A IC50 estimated: ~80.51 µM (24 h) |
||||
| Breast cancer | MCF-7 MDA-MB-231 H184B5F5/M10 |
Effective conc: 25 µM and 50 µM (48 h) Tumor cell lines: MCF-7 MDA-MB-231 |
|
[65] |
| Normal cell line: H184B5F5/M10 (ISL did not influence the viability) |
||||
| MCF-7 MCF-7/ADR MCF-10A |
Tumor cell lines: MCF-7 IC50 estimation: ~59.39 µM MCF-7/ADR IC50 estimation: ~38.86 µM (24 h) |
|
[53] | |
| Normal cell line: MCF-10A ISL (at 100 µM) had limited inhibitory effects on the proliferation |
||||
| MDA-MB-231 | Testing conc.: 0, 10, 25, 50 µM MDA-MB-231 IC50 estimated: ~24.23 µM (48 h) |
|
[66] | |
| MCF-7aro | Testing conc.: 0, 0.625, 1.25, 2.5, 5, 10 µM MCF-7aro IC50: 2.5 µM (24 h) |
|
[44] | |
| Colon cancer | HT29 | HT29 ED50: 11.1 µg/mL (42.32 µM) |
|
[67] |
| HT29 | Testing conc.: 0, 5,10, 20, 30, 40, 50 µM 40 µM was applied; (24 h) |
|
[68] | |
| HCT116 HT29 SW480 |
Testing conc.: 0,10, 20, 30, 40 µM HCT116 IC50 estimated = ~42.41 µM Working conc.: 30 or 40 µM; (24 h) |
|
[69] | |
| HCT116 | Testing Conc.: 0, 2.5,5, 10, 20, 40, 80, 160 µM HCT116 IC50 estimated: ~78.78 µM (48 h) HCT116 IC50 estimated: ~53.97 µM (72 h) HCT116 IC50 estimated: ~44.8 µM (96 h) |
|
[70] | |
| CT26 | Testing Conc.: 0, 10, 20, 40, 60, 80 µM CT26 IC50 estimated = ~54.48 µM |
|
[71] | |
| Colon26 RCN9 CoLo-320DM |
Testing Conc.: 0, 5, 25, 100 µM (24, 48 h) Colon26 IC50 estimated = ~17.55 µM (24 h) Colon26 IC50 estimated = ~12.59 µM (48 h) RCN9 IC50 estimated = ~41.73 µM (24 h) RCN9 IC50 estimated = ~18.21 µM (48 h) CoLo-320DM IC50 estimated = ~23.10 µM (24 h) CoLo-320DM IC50 estimated = ~10.82 µM (48 h) |
|
[72] | |
| Colon cancer | HCT116 | Applied 20 µM (48 h) |
|
[73] |
| Caco-2/TC-7 | Caco-2/TC-7 EC50: 42 μM |
|
[74] | |
| Ovary cancer | SKOV3 OVCAR5 ES2 |
Testing conc.: 2, 4, 8, 16, 32, 64, and 100 µM SKOV3 IC50: 83.2 µM (72 h) OVCAR5 IC50: 55.5 µM (72 h) ES2 IC50: 40.1 µM (72 h) Effective Conc.: 10 µM |
|
[75] |
| SKOV3 OVCAR5 |
Testing conc.: 0, 1, 5, 10, 20, 25, 50, 75, and 100 µM OVCAR5 IC50: 11 µM (48 h) ES2 IC50: 25 µM (48 h) |
|
[76] | |
| Antral follicle culture (female CD-1 mic) | Testing conc.: 0.6, 6, 36, and 100 μM |
|
[77] | |
| SKOV3 OVCAR3 | Testing conc.: 5~80 μM 30 μM applied |
|
[78] | |
| SKOV3 | N.A. |
|
[76][79] | |
| Lung cancer | H1299 H1975 A549 |
H1299 IC50 estimated: ~36.78~46.08 µM H1975 IC50: 48.14 µM A549 IC50: 75.08 µM (48 h) |
|
[9] |
| A549 | A549: applied 20 µM (24 h) |
|
[80][81] | |
| RAW 264.7 | Testing conc.: 5, 10, 20 µM for (Pretreated with 10mM of t-BHP for 18 h) RAW 264.7 (treated with t-BHP) EC50 = 10 µM (18 h) |
|
[82] | |
| Calu-3 | Calu-3 cells were infected with PR8/H1N1 virus; [EC50] = 24.7 μM |
|
[83] | |
| H1650 H1975 A549 |
H1650 IC50 estimated: ~26.88 µM (24 h) H1975 IC50 estimated: ~8.92 µM (24 h) A549 IC50 estimated: ~46.7 µM (24 h) |
|
[38] | |
| A549 | A549 IC50: 0.05 mg/mL (~191.21 µM ~117 µM) |
|
[84] | |
| Lung cancer | guinea-pig tracheal smooth muscle | N.A. |
|
[85] |
| A549 | A549 IC50: 27.14 µM |
|
[86] | |
| A549 | A549 IC50: 18.5 µM |
|
[87] | |
| AML (acute myeloid leukemia) |
HL-60 | HL-60 ED50: 5.5 µg/mL (~21.46 µM) 5.00 µg/mL = 19.5 µM (72 h) |
|
[61] |
| MV4-11 MOLM-13 OCI-LY10 |
MV4-11 IC50: 3.2 + 1.2 µM; MOLM-13 IC50: 4.9 + 2.1 µM OCI-LY10 IC50: 20.1 ± 6.7 µM (72 h) |
|
[31] | |
| LCLs | Testing conc.: 0, 20, 40, 60, 80, 100, 120, 140 µM LCLs IC50 estimated: 40~65 µM (24 h) Applied 50 µM for studies. |
|
[80] | |
| HL-60 | Testing conc.: 1~15 µg/mL (3.9 µM~58.54 µM) HL-60 IC50 estimated: ~40.42 µM (72 h) |
|
[81] | |
| RAW264.7 | Testing conc.: 20 and 50 μM |
|
[88] | |
| AML (acute myeloid leukemia) |
RAW264.7 | Testing conc.: 50 and 100 μM |
|
[89] |
| HL-60 | Testing conc.: 2.5~20 μg/mL (3.9 µM~78.05 µM) (Working conc.: 72 µM) |
|
[90] | |
| HL-60 | Testing conc.: 2.5~10 μg/mL (3.9 µM~39.0 µM) |
|
[91] | |
| Jurkat J-Jhan J16 HUT78 Karpas 45 |
Jurkat IC50: 0.49 ± 0.12 nM (72 h) J-Jhan IC50: 1.55 ± 1.12 nM (72 h) J16 IC50: 5.25 ± 1.12 µM (72 h) HUT78 IC50: 11 ±13.5 µM (72 h) Karpas 45 IC50: 6.61 ± 1.07 µM (72 h) |
|
[92] | |
| CCRF-CEM | CCRF-CEM IC50: 18.38 μM (24~72 h) |
|
[93] | |
| AML (acute myeloid leukemia) |
Human monocyte model THP-1 | N.A. |
|
[94] |
| Melanoma | A375 A2058 |
Testing Conc: 0, 10, 20, 40, 80 µM A375 IC50: 21.63 µM (24 h) A2058 IC50: 20.75 µM (24 h) |
|
[95] |
| B16F0 | N.A. |
|
[96] | |
| A375 | Testing Conc.: 0, 5, 10, 15 μg/mL (15 μg/mL = 58.53 µM) A375 IC50 estimated: ~48 µM |
|
[97] | |
| A375 | 40 μg/mL: 69.86% 60 μg/mL: 92.22% A375 IC50 estimated: ~73 µM (24 h) |
|
[98] | |
| Melanoma | B16F0 | Testing Conc.: 20, 40, 60 and 80 μg/mL B16F10 IC50 estimated: 35 μg/mL (~41.576 μM; 24 h) B16F10 IC50 estimated: 22 μg/mL (~86.77 μM; 48 h) |
|
[99] |
| B16F10 | Testing Conc.: 5, 10, 15, 20, and 25 μg/mL B16F10 IC50 estimated: ~19 μg/mL (~74.595 μM; 24 h) B16F10 IC50 estimated: ~10.5 μg/mL (~41.576 μM; 48 h) |
|
[100] | |
| ARH-77 U266 MPC-11 SP2/0 CZ-1 RPMI8226 |
ARH-77 IC50: ~13.54 µM MPC-11 IC50: ~4.45 µM SP2/0 IC50: ~22.91 µM CZ-1 IC50: ~13.93 µM U266 IC50: ~8.62 µM RPMI8226 IC50: ~9.09 µM IC50 of ISL was < 4 μg/mL (48 h) |
|
[101] | |
| SK-MEL-2 HaCaT |
Testing Conc.: 0, 1, 4, and 8 µM SK-MEL-2 cells and HaCaT cells (48 h) treated less than 8 µM showed no cytotoxic effects |
|
[102] | |
| Melanoma | B16 mouse melanoma 4A5 cells | Testing 150 and 200 µM (18 and 24 h) |
|
[103] |
| HCC/Hepato-ma | Hep3B | Hep3B IC50: 42.84 + 2.01 μM 50 μM applied (48 h) |
|
[104] |
| HepG2 Hep3B |
Testing conc.: 20, 40, 60, 80, and 100 μM (18 h) HepG2 IC50: 27.71 μM Hep3B IC50: 35.28 μM |
|
[105] | |
| HepG2 | Testing conc.: 1, 5, 10, 20 μg HepG2 IC50 estimated: ~88.46 μM (24 h) HepG2 IC50 estimated: ~31.07 μM (48 h) |
|
[106] | |
| HepG2 | HepG2 IC50: 10.51 μg/mL (~39 μM; 48 h) |
|
[107] | |
| HCC/Hepato-ma | SNU475 | SNU475 IC50: 0.243 + 0.21 mM |
|
[58] |
| Hepa 1c1c7 | Hepa 1c1c7 IC50: 36.3 μM |
|
[108] | |
| Hep3B | Hep3B IC50: 50.8 μM |
|
[43] | |
| SK-Hep-1 | SK-Hep-1 IC50: 19.08 μM |
|
[109] | |
| PC-3 22RV1 |
Testing conc: 0, 1, 10, 25, 50, and 100 μM) PC-3 IC50: 19.6 μM (48 h) 22RV1 IC50: 36.6 μM (48 h) |
|
[110] | |
| Prostate cancer | C4-2 LNCaP IEC-6 |
10~100 μM (24 h) C4-2 IC50: 87.0 μM |
|
[59] |
| DU145 | Applied conc.: 5~20 μM |
|
[111] | |
| DU145 | Applied conc.: 0~20 μM |
|
[112] | |
| DU145 | Applied conc.: 0~20 μM |
|
[113] | |
| Prostate cancer | MAT-LyLu DU145 |
Applied conc.: 0~20 μM MAT-LyLuIC50 estimated: ~13.74/5.67/5.01 µM DU145 IC50 estimated: ~56.87/31.49/17.60 µM (24 h/48 h/72 h) |
|
[114] |
| DU145 LNCaP | Testing conc.: 0, 5, 10, 15, and 20 μM DU145 IC50 estimated: ~10.561 µM (48 h) LNCaP IC50 estimated: ~10.775 µM (48 h) |
|
[115] | |
| Cervical cancer | Ca Ski SiHa HeLa C-33A |
Testing conc: 10, 20, 40, and 80 µM Ca Ski IC50 estimated: 39.09 μM (72 h) SiHa IC50 estimated: 53.76 μM (72 h) HeLa IC50 estimated: 58.10 μM (72 h) C-33A IC50 estimated: 32.83 μM (72 h) |
|
[116] |
| HeLa | Testing conc: 2, 5, 10, 30, 40, and 60 μg/mL HeLa IC50 estimated: ~21.24 μM (24 h) |
|
[117] | |
| HeLa | HeLa IC50: 9.8 μM (48 h) |
|
[118] | |
| Gastric cancer | MKN28 | MKN28 IC50: ~20.84 µM (48 h) |
|
[119] |
| MKN-45 | 5 µM applied |
|
[120] | |
| MGC-803 | 0.11 g/L applied (24 h) |
|
[121] | |
| SGC-7901 BGC-823 | BGc-823 IC50: 23.18 µM (48 h) SGC-7901 IC50: 12.91 µM (48 h) |
|
[34] | |
| Uterine leiomyoma | Leiomyma Myomentrium |
Testing conc: 0, 10, 20, 50 µM Leiomyma IC50 estimated = ~39.33 µM Myomentrium IC50 estimated = ~698.8 µM (48 h) |
|
[122] |
| Osteosarcoma | U2OS | Testing conc: 5, 10, and 20 µM 20 μM applied |
|
[123][128] |
| Saos‑2 MC3T3-E1 |
Saos‑2 IC50 estimated = ~24.23 μM 30 μM applied |
|||
| Glioma | SK-N-BE(2) IMR-32 | Effective conc. > 5 µM |
|
[125] |
| U87 | U87 IC50: 6.3 µM |
|
[124] | |
| PC12 | PC12 IC50: 17.8 ± 1.8 μM |
|
[126] | |
| Bladder cancer | T24 | Effective conc.: 30 and 70 µg/mL (24 h) |
|
[127] |
| Oral squamous cell carcinomas (OSCC) |
SG SAS-CSCs OECM-1 |
SG cells IC50: 386.3 ± 29.7 μM SAS-CSCs IC50: 144.9 ± 25.7 μM OECM-1-CSCs IC50: 104.5 ± 26.2 μM |
|
[54] |
ote: The ‘’IC50 estimated’’ indicated Data extracted from published figures using Web Plot Digitizer (https://automeris.io/WebPlotDigitizer), then analyzed IC50 by “Quest Graph™ IC50 Calculator.” AAT Bioquest Inc, 27 October 2020, https://www.aatbio.com/tools/ic50-calculator [133].
References
- Guo, J.; Liu, D.; Nikolic, D.; Zhu, D.; Pezzuto, J.M.; Van Breemen, R.B. In Vitro Metabolism of Isoliquiritigenin by Human Liver Microsomes. Drug Metab. Dispos. 2007, 36, 461–468, doi:10.1124/dmd.107.018721.
- Cuendet, M.; Guo, J.; Luo, Y.; Chen, S.-N.; Oteham, C.P.; Moon, R.C.; Van Breemen, R.B.; Marler, L.E.; Pezzuto, J.M. Cancer Chemopreventive Activity and Metabolism of Isoliquiritigenin, a Compound Found in Licorice. Cancer Prev. Res. 2010, 3, 221–232, doi:10.1158/1940-6207.CAPR-09-0049.
- Guo, J.; Liu, A.; Cao, H.; Luo, Y.; Pezzuto, J.M.; Van Breemen, R.B. Biotransformation of the Chemopreventive Agent 2′,4′,4-Trihydroxychalcone (Isoliquiritigenin) by UDP-Glucuronosyltransferases. Drug Metab. Dispos. 2008, 36, 2104–2112, doi:10.1124/dmd.108.021857.
- Yang, E.-J.; Kim, M.; Woo, J.E.; Lee, T.; Jung, J.-W.; Song, K.-S. The comparison of neuroprotective effects of isoliquiritigenin and its Phase I metabolites against glutamate-induced HT22 cell death. Bioorg. Med. Chem. Lett. 2016, 26, 5639–5643, doi:10.1016/j.bmcl.2016.10.072.
- Chen, C.; Shenoy, A.K.; Padia, R.; Fang, D.-D.; Jing, Q.; Yang, P.; Shi-Bing, S.; Huang, S. Suppression of lung cancer progres-sion by isoliquiritigenin through its metabolite 2, 4, 2’, 4’-Tetrahydroxychalcone. J. Exp. Clin. Cancer Res. 2018, 37, 243, doi:10.1186/s13046-018-0902-4.
- Lee, Y.K.; Chin, Y.-W.; Bae, J.-K.; Seo, J.S.; Choi, Y.H. Pharmacokinetics of Isoliquiritigenin and Its Metabolites in Rats: Low Bioavailability Is Primarily Due to the Hepatic and Intestinal Metabolism. Planta Med. 2013, 79, 1656–1665, doi:10.1055/s-0033-1350924.
- Zhou, J.-X.; Wink, M. Reversal of Multidrug Resistance in Human Colon Cancer and Human Leukemia Cells by Three Plant Extracts and Their Major Secondary Metabolites. Medicines 2018, 5, 123, doi:10.3390/medicines5040123.
- Yushan, R.; Ying, Y.; Yujun, T.; Jingchun, Y.; Dongguang, L.; Lihong, P.; Pingping, W.; Lili, Z.; Fanhui, Z.; Zhong, L.; et al. Isoliquiritigenin inhibits mouse S180 tumors with a new mechanism that regulates autophagy by GSK-3beta/TNF-alpha pathway. Eur. J. Pharmacol. 2018, 838, 11–22.
- Han, B.; Chen, W.; Zheng, Q.; Wang, X.; Yan, H.; Li, L.; Aisa, H. Determination of Isoliquiritigenin and Its Distribution in Mice by Synchronous Fluorescence Spectrometry. Anal. Sci. 2011, 27, 1115, doi:10.2116/analsci.27.1115.
- Qiao, H.; Zhang, X.; Wang, T.; Liang, L.; Chang, W.; Xia, H. Pharmacokinetics, biodistribution and bioavailability of isoliquiritigenin after intravenous and oral administration. Pharm. Biol. 2014, 52, 228–236, doi:10.3109/13880209.2013.832334.
- Li, H.; Ye, M.; Zhang, Y.; Huang, M.; Xu, W.; Chu, K.; Chen, L.; Que, J. Blood-brain barrier permeability of Gualou Guizhi granules and neuroprotective effects in ischemia/reperfusion injury. Mol. Med. Rep. 2012, 12, 1272–1278, doi:10.3892/mmr.2015.3520.
- Ma, X.; Fang, F.; Song, M.; Ma, S. The effect of isoliquiritigenin on learning and memory impairments induced by high-fat diet via inhibiting TNF-α/JNK/IRS signaling. Biochem. Biophys. Res. Commun. 2015, 464, 1090–1095, doi:10.1016/j.bbrc.2015.07.081.
- Zhang, K.; Wang, Q.; Yang, Q.; Wei, Q.; Man, N.; Adu-Frimpong, M.; Toreniyazov, E.; Ji, H.; Yu, J.; Xu, X. Enhancement of Oral Bioavailability and Anti-hyperuricemic Activity of Isoliquiritigenin via Self-Microemulsifying Drug Delivery System. AAPS PharmSciTech 2019, 20, 218, doi:10.1208/s12249-019-1421-0.
- Liu, J.; Wang, Q.; Adu-Frimpong, M.; Wei, Q.; Xie, Y.; Zhang, K.; Wei, C.; Weng, W.; Ji, H.; Toreniyazov, E.; et al. Prepara-tion, in vitro and in vivo evaluation of isoliquiritigenin-loaded TPGS modified proliposomes. Int. J. Pharm. 2019, 563, 53–62, doi:10.1016/j.ijpharm.2019.03.034.
- Zhang, X.; Zhang, T.; Shi, Y.; Ni, J. Enhancement of gastrointestinal absorption of isoliquiritigenin by nanostructured lipid carrier. Adv. Powder Technol. 2014, 25, 1060–1068.
- Qiao, F.; Zhao, Y.; Mai, Y.; Guo, J.; Dong, L.; Zhang, W.; Yang, J. Isoliquiritigenin Nanosuspension Enhances Cytostatic Ef-fects in A549 Lung Cancer Cells. Planta Med. 2020, 86, 538–547, doi:10.1055/a-1134-3378.
- Sun, X.; Zhang, J.; Wang, Z.; Liu, B.; Zhu, S.; Zhu, L.; Peng, B. Licorice isoliquiritigenin-encapsulated mesoporous silica na-noparticles for osteoclast inhibition and bone loss prevention. Theranostics 2019, 9, 5183–5199, doi:10.7150/thno.33376.
- Gao, F.; Zhang, J.; Fu, C.; Xie, X.; Peng, F.; You, J.; Tang, H.; Wang, Z.; Li, P.; Chen, J. iRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int. J. Nanomed. 2017, 12, 4147–4162.
- Zhang, X.; Qiao, H.; Chen, Y.; Li, L.; Xia, H.; Shi, Y. Preparation, properties and preclinical pharmacokinetics of low molec-ular weight heparin-modified isoliquiritigenin-loaded solid lipid nanoparticle. Iran. J. Pharm. Res. 2016, 15, 269–282.
- Zhang, X.-Y.; Qiao, H.; Ni, J.M.; Shi, Y.B.; Qiang, Y. Preparation of isoliquiritigenin-loaded nanostructured lipid carrier and the in vivo evaluation in tumor-bearing mice. Eur. J. Pharm. Sci. 2013, 49, 411–422, doi:10.1016/j.ejps.2013.04.020.
- Noh, G.Y.; Suh, J.Y.; Park, S.N. Ceramide-based nanostructured lipid carriers for transdermal delivery of isoliquiritigenin: Development, physicochemical characterization, and in vitro skin permeation studies. Korean J. Chem. Eng. 2017, 34, 400–406, doi:10.1007/s11814-016-0267-3.
- Xie, Y.-J.; Wang, Q.; Adu-Frimpong, M.; Liu, J.; Zhang, K.-Y.; Xu, X.; Yu, J.-N. Preparation and evaluation of isoliquiritigen-in-loaded F127/P123 polymeric micelles. Drug Dev. Ind. Pharm. 2019, 45, 1224–1232, doi:10.1080/03639045.2019.1574812.
- Wang, G.; Yu, Y.; Wang, Y.-Z.; Yin, P.-H.; Xu, K.; Zhang, H. The effects and mechanisms of isoliquiritigenin loaded nanolip-osomes regulated AMPK/mTOR mediated glycolysis in colorectal cancer. Artif. Cells Nanomed. Biotechnol. 2020, 48, 1231–1249, doi:10.1080/21691401.2020.1825092.
- Kong, B.J.; Kim, A.; Park, S.N. Properties and in vitro drug release of hyaluronic acid-hydroxyethyl cellulose hydrogels for transdermal delivery of isoliquiritigenin. Carbohydr. Polym. 2016, 147, 473–481, doi:10.1016/j.carbpol.2016.04.021.
- Cao, M.; Zhan, M.; Wang, Z.; Wang, Z.; Li, X.-M.; Miao, M. Development of an Orally Bioavailable Isoliquiritigenin Self-Nanoemulsifying Drug Delivery System to Effectively Treat Ovalbumin-Induced Asthma. Int. J. Nanomed. 2020, 15, 8945–8961, doi:10.2147/ijn.s269982.
- Jing, Z.; Ji-Liang, W.; Lin, Z.; Chun, Z. Preparation of isoliquiritigenin liposome and its inhibitive effects on proliferation of human cervical cancer cells in vitro. Chin. J. Clin. Pharmacol. Ther. 2004, 11, 1268–1272.
- Peng, F.; Meng, C.-W.; Zhou, Q.-M.; Chen, J.-P.; Xiong, L. Cytotoxic Evaluation against Breast Cancer Cells of Isoliquiriti-genin Analogues from Spatholobus suberectus and Their Synthetic Derivatives. J. Nat. Prod. 2015, 79, 248–251, doi:10.1021/acs.jnatprod.5b00774.
- Huang, F.; Wang, J.; Xu, Y.; Zhang, Y.; Xu, N.; Yin, L. Discovery of novel isoliquiritigenin analogue ISL-17 as a potential anti-gastric cancer agent. Biosci. Rep. 2020, 40, 20201199, doi:10.1042/bsr20201199.
- Peng, F.; Xiong, L.; Xie, X.; Tang, H.; Huang, R.; Peng, C. Isoliquiritigenin Derivative Regulates miR-374a/BAX Axis to Sup-press Triple-Negative Breast Cancer Tumorigenesis and Development. Front. Pharmacol. 2020, 11, 378, doi:10.3389/fphar.2020.00378.
- Jeong, S.; Lee, S.; Kim, K.; Lee, Y.; Lee, J.; Oh, S.; Choi, J.W.; Kim, S.W.; Hwang, K.C.; Lim, S. Isoliquiritigenin Derivatives inhibit RANKL-induced osteoclastogenesis by regulating p38 and NF-κB activation in RAW 264.7 cells. Molecules 2020, 25, 3908.
- Gaur, R.; Yadav, K.S.; Verma, R.K.; Yadav, N.P.; Bhakuni, R.S. In vivo anti-diabetic activity of derivatives of isoliquiritigen-in and liquiritigenin. Phytomedicine 2014, 21, 415–422, doi:10.1016/j.phymed.2013.10.015.
- Selvaraj, B.; Kim, D.W.; Huh, G.; Lee, H.; Kang, K.; Lee, J.W. Synthesis and biological evaluation of isoliquiritigenin deriva-tives as a neuroprotective agent against glutamate mediated neurotoxicity in HT22 cells. Bioorg. Med. Chem. Lett. 2020, 30, 127058, doi:10.1016/j.bmcl.2020.127058.
- Reddy, M.R.; Aidhen, I.S.; Reddy, U.A.; Reddy, G.B.; Ingle, K.; Mukhopadhyay, S.; Ingle, K. Synthesis of 4-C -β-D-Glucosylated Isoliquiritigenin and Analogues for Aldose Reductase Inhibition Studies. Eur. J. Org. Chem. 2019, 2019, 3937–3948, doi:10.1002/ejoc.201900413
- Gay, N.H.; Suwanjang, W.; Ruankham, W.; Songtawee, N.; Wongchitrat, P.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Butein, isoliquiritigenin, and scopoletin attenuate neurodegeneration via antioxidant enzymes and SIRT1/ADAM10 sig-naling pathway. RSC Adv. 2020, 10, 16593–16606, doi:10.1039/c9ra06056a.
- Wang, Z.; Wang, N.; Han, S.; Wang, D.; Mo, S.; Yu, L.; Huang, H.; Tsui, K.; Shen, J.; Chen, J. Dietary Compound Isoliquiriti-genin Inhibits Breast Cancer Neoangiogenesis via VEGF/VEGFR-2 Signaling Pathway. PLoS ONE 2013, 8, e68566, doi:10.1371/journal.pone.0068566.
- Wang, N.; Wang, Z.; Peng, C.; You, J.; Shen, J.; Han, S.; Chen, J. Dietary compound isoliquiritigenin targets GRP78 to chemosensitize breast cancer stem cells via beta-catenin/ABCG2 signaling. Carcinogenesis 2014, 35, 2544–2554.
- Cao, Z.-X.; Wen, Y.; He, J.-L.; Huang, S.-Z.; Gao, F.; Guo, C.-J.; Liu, Q.-Q.; Zheng, S.-W.; Gong, D.-Y.; Li, Y.-Z.; et al. Isoliquiri-tigenin, an Orally Available Natural FLT3 Inhibitor from Licorice, Exhibits Selective Anti-Acute Myeloid Leukemia Efficacy In Vitro and In Vivo. Mol. Pharmacol. 2019, 96, 589–599, doi:10.1124/mol.119.116129.
- Jung, S.K.; Lee, M.-H.; Lim, D.Y.; Kim, J.E.; Singh, P.; Lee, S.-Y.; Jeong, C.-H.; Lim, T.-G.; Chen, H.; Chi, Y.-I.; et al. Isoliquiri-tigenin Induces Apoptosis and Inhibits Xenograft Tumor Growth of Human Lung Cancer Cells by Targeting Both Wild Type and L858R/T790M Mutant EGFR. J. Biol. Chem. 2014, 289, 35839–35848, doi:10.1074/jbc.m114.585513.
- Yan, F.; Yang, F.; Wang, R.; Yao, X.J.; Bai, L.; Zeng, X.; Huang, J.; Wong, V.K.; Lam, C.W.; Zhou, H.; et al. Isoliquiritigenin suppresses human T Lymphocyte activation via covalently binding cysteine 46 of IkappaB kinase. Oncotarget 2017, 8, 34223–34235.
- Park, S.J.; Youn, H.S. Isoliquiritigenin suppresses the Toll-interleukin-1 receptor domain-containing adapter inducing in-terferon-beta (TRIF)-dependent signaling pathway of Toll-like receptors by targeting TBK1. J. Agric. Food Chem. 2010, 58, 4701–4705.
- Qi, X.; Zhang, N.; Zhao, L.; Hu, L.; Cortopassi, W.A.; Jacobson, M.P.; Li, X.; Zhong, R. Structure-based identification of novel CK2 inhibitors with a linear 2-propenone scaffold as anti-cancer agents. Biochem. Biophys. Res. Commun. 2019, 512, 208–212, doi:10.1016/j.bbrc.2019.03.016.
- Kim, D.-C.; Choi, S.-Y.; Kim, S.-H.; Yun, B.-S.; Yoo, I.-D.; Reddy, N.R.P.; Yoon, H.S.; Kim, K.-T. Isoliquiritigenin Selectively Inhibits H2 Histamine Receptor Signaling. Mol. Pharmacol. 2006, 70, 493–500, doi:10.1124/mol.106.023226.
- Wang, C.; Chen, L.; Cai, Z.C.; Chen, C.; Liu, Z.; Liu, X.; Zou, L.; Chen, J.; Tan, M.; Wei, L.; et al. Comparative Proteomic Analysis Reveals the Molecular Mechanisms Underlying the Accumulation Difference of Bioactive Constituents in Glycyr-rhiza uralensis Fisch under Salt Stress. J. Agric. Food Chem. 2020, 68, 1480–1493, doi:10.1021/acs.jafc.9b04887.
- Khan, S.I.; Zhao, J.; Ibrahim, M.; Walker, L.A.; DasMahapatra, A.K. Potential utility of natural products as regulators of breast cancer-associated aromatase promoters. Reprod. Biol. Endocrinol. 2011, 9, 91, doi:10.1186/1477-7827-9-91.
- Shah, U.; Patel, S.; Patel, M.; Gandhi, K.; Patel, A. Identification of chalcone derivatives as putative non-steroidal aromatase inhibitors potentially useful against breast cancer by molecular docking and ADME prediction. Indian J. Chem. Sect. B 2020, 59, 283–293.
- Li, Z.-X.; Li, J.; Li, Y.; You, K.; Xu, H.; Wang, J. Novel insights into the apoptosis mechanism of DNA topoisomerase I inhib-itor isoliquiritigenin on HCC tumor cell. Biochem. Biophys. Res. Commun. 2015, 464, 548–553, doi:10.1016/j.bbrc.2015.07.003.
- Wang, N.; Wang, Z.; Wang, Y.; Xie, X.; Shen, J.; Peng, C.; You, J.; Peng, F.; Tang, H.; Guan, X.; et al. Dietary compound isoliquiritigenin prevents mammary carcinogenesis by inhibiting breast cancer stem cells through WIF1 demethylation. Oncotarget 2015, 6, 9854–9876, doi:10.18632/oncotarget.3396.
- Kong, L.D.; Zhang, Y.; Pan, X.; Tan, R.X.; Cheng, C.H.K. Inhibition of xanthine oxidase by liquiritigenin and isoliquiriti-genin isolated from Sinofranchetia chinensis. Cell. Mol. Life Sci. 2000, 57, 500–505, doi:10.1007/pl00000710.
- Wu, C.-H.; Chen, H.-Y.; Wang, C.-W.; Shieh, T.-M.; Huang, T.-C.; Lin, L.-C.; Wang, K.-L.; Hsia, S.-M. Isoliquiritigenin induces apoptosis and autophagy and inhibits endometrial cancer growth in mice. Oncotarget 2016, 7, 73432–73447, doi:10.18632/oncotarget.12369.
- Na, A.-Y.; Yang, E.-J.; Jeon, J.M.; Ki, S.H.; Song, K.-S.; Lee, S. Protective Effect of Isoliquiritigenin against Ethanol-Induced Hepatic Steatosis by Regulating the SIRT1-AMPK Pathway. Toxicol. Res. 2018, 34, 23–29, doi:10.5487/TR.2018.34.1.023.
- Younas, M.; Hano, C.; Giglioli-Guivarc’H, N.; Abbasi, B.H. Mechanistic evaluation of phytochemicals in breast cancer rem-edy: Current understanding and future perspectives. RSC Adv. 2018, 8, 29714–29744, doi:10.1039/c8ra04879g.
- Wang, Z.; Wang, N.; Liu, P.; Chen, Q.; Situ, H.; Xie, T.; Zhang, J.; Peng, C.; Lin, Y.; Chen, J. MicroRNA-25 regulates chemo-resistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiriti-genin. Oncotarget 2014, 5, 7013–7026, doi:10.18632/oncotarget.2192.
- Wang, K.-L.; Hsia, S.-M.; Chan, C.-J.; Chang, F.-Y.; Huang, C.-Y.; Bau, D.-T.; Wang, P.S. Inhibitory effects of isoliquiritigenin on the migration and invasion of human breast cancer cells. Expert Opin. Ther. Targets 2013, 17, 337–349, doi:10.1517/14728222.2013.756869.
- Kwon, H.M.; Choi, Y.J.; Choi, J.S.; Kang, S.W.; Bae, J.Y.; Kang, I.J.; Jun, J.G.; Lee, S.S.; Lim, S.S.; Kang, Y.H. Blockade of cyto-kine-induced endothelial cell adhesion molecule expression by licorice isoliquiritigenin through NF-kappaB signal disrup-tion. Exp. Biol. Med. 2007, 232, 235–245.
- Zhang, X.; Yeung, E.D.; Wang, J.; Panzhinskiy, E.E.; Tong, C.; Li, W.; Li, J. Isoliquiritigenin, a natural anti-oxidant, selective-ly inhibits the proliferation of prostate cancer cells. Clin. Exp. Pharmacol. Physiol. 2010, 37, 841–847.
- Hu, F.-W.; Yu, C.-C.; Hsieh, P.-L.; Liao, Y.-W.; Lu, M.-Y.; Chu, P.-M. Targeting oral cancer stemness and chemoresistance by isoliquiritigenin-mediated GRP78 regulation. Oncotarget 2017, 8, 93912–93923, doi:10.18632/oncotarget.21338.
- Maggiolini, M.; Statti, G.; Vivacqua, A.; Gabriele, S.; Rago, V.; Loizzo, M.; Menichini, F.; Andò, S. Estrogenic and antiprolif-erative activities of isoliquiritigenin in MCF7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2002, 82, 315–322, doi:10.1016/s0960-0760(02)00230-3.
- Anemone, A.; Consolino, L.; Conti, L.; Reineri, F.; Cavallo, F.; Aime, S.; Longo, D.L. In vivo evaluation of tumour acidosis for assessing the early metabolic response and onset of resistance to dichloroacetate by using magnetic resonance pH im-aging. Int. J. Oncol. 2017, 51, 498–506, doi:10.3892/ijo.2017.4029.
- Lau, G.T.Y.; Ye, L.; Leung, L.K. The Licorice Flavonoid Isoliquiritigenin Suppresses Phorbol Ester-induced Cyclooxygen-ase-2 Expression in the Non-tumorigenic MCF-10A Breast Cell Line. Planta Med. 2009, 76, 780–785, doi:10.1055/s-0029-1240699.
- Zheng, H.; Li, Y.; Wang, Y.; Zhao, H.; Zhang, J.; Chai, H.; Tang, T.; Yue, J.; Guo, A.M.; Yang, J. Downregulation of COX-2 and CYP 4A signaling by isoliquiritigenin inhibits human breast cancer metastasis through preventing anoikis resistance, mi-gration and invasion. Toxicol. Appl. Pharmacol. 2014, 280, 10–20, doi:10.1016/j.taap.2014.07.018.
- Ning, S.; Mu, J.; Shen, Z.; Zhu, D.; Jiang, F.; Wang, X.; Li, Y. Isoliquiritigenin attenuates the invasive capacity of breast cancer cells via up-regulating the tumor suppressor RECK. RSC Adv. 2016, 6, 24719–24727, doi:10.1039/C5RA26759E.
- Li, Y.; Zhao, H.; Wang, Y.; Zheng, H.; Yu, W.; Chai, H.; Zhang, J.; Falck, J.R.; Guo, A.M.; Yue, J.; et al. Isoliquiritigenin induc-es growth inhibition and apoptosis through downregulating arachidonic acid metabolic network and the deactivation of PI3K/Akt in human breast cancer. Toxicol. Appl. Pharmacol. 2013, 272, 37–48, doi:10.1016/j.taap.2013.05.031.
- Peng, F.; Tang, H.; Liu, P.; Shen, J.; Guan, X.-Y.; Xie, X.; Gao, J.; Xiong, L.; Jiangang, S.; Chen, J.; et al. Isoliquiritigenin modu-lates miR-374a/PTEN/Akt axis to suppress breast cancer tumorigenesis and metastasis. Sci. Rep. 2017, 7, 1–14, doi:10.1038/s41598-017-08422-y.
- Ning, S.; Zhu, D.; Shen, Z.; Liu, J.; Liu, Y.; Chen, J.; Li, Z. Isoliquiritigenin attenuates MiR-21 expression via induction of PI-AS3 in breast cancer cells. RSC Adv. 2017, 7, 18085–18092.
- Wang, Z.; Wang, N.; Liu, P.; Chen, Q.; Situ, H.; Xie, T.; Zhang, J.; Peng, C.; Lin, Y.; Chen, J. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget 2014, 5, 7013–7026, doi:10.18632/oncotarget.2192.
- Lin, P.-H.; Chiang, Y.-F.; Shieh, T.-M.; Chen, H.-Y.; Shih, C.-K.; Wang, T.-H.; Wang, K.-L.; Huang, T.-C.; Hong, Y.-H.; Li, S.-C.; et al. Dietary Compound Isoliquiritigenin, an Antioxidant from Licorice, Suppresses Triple-Negative Breast Tumor Growth via Apoptotic Death Program Activation in Cell and Xenograft Animal Models. Antioxidants 2020, 9, 228, doi:10.3390/antiox9030228.
- Zorko, B.A.; Pérez, L.B.; De Blanco, E.J.C. Effects of ILTG on DAPK1 promoter methylation in colon and leukemia cancer cell lines. Anticancer. Res. 2010, 30, 3945–3950.
- Yoshida, T.; Horinaka, M.; Takara, M.; Tsuchihashi, M.; Mukai, N.; Wakada, M.; Sakai, T. Combination of isoliquiritigenin and tumor necrosis factor-related apoptosis-inducing ligand induces apoptosis in colon cancer HT29 cells. Environ. Health Prev. Med. 2008, 13, 281–287, doi:10.1007/s12199-008-0041-1.
- Jin, H.; Lee, S.H.; Lee, S.H. Isoliquiritigenin-mediated p62/SQSTM1 induction regulates apoptotic potential through attenua-tion of caspase-8 activation in colorectal cancer cells. Eur. J. Pharmacol. 2018, 841, 90–97, doi:10.1016/j.ejphar.2018.10.015.
- Auyeung, K.K.-W.; Auyeung, J.K.K.A.K.K. Novel herbal flavonoids promote apoptosis but differentially induce cell cycle arrest in human colon cancer cell. Investig. New Drugs 2009, 28, 1–13, doi:10.1007/s10637-008-9207-3.
- Lee, C.K.; Son, S.H.; Park, K.K.; Park, J.H.Y.; Lim, S.S.; Chung, W.-Y. Isoliquiritigenin Inhibits Tumor Growth and Protects the Kidney and Liver Against Chemotherapy-Induced Toxicity in a Mouse Xenograft Model of Colon Carcinoma. J. Phar-macol. Sci. 2008, 106, 444–451, doi:10.1254/jphs.fp0071498.
- Takahashi, T.; Takasuka, N.; Iigo, M.; Baba, M.; Nishino, H.; Tsuda, H.; Okuyama, T. Isoliquiritigenin, a flavonoid from lico-rice, reduces prostaglandin E2 and nitric oxide, causes apoptosis, and suppresses aberrant crypt foci development. Cancer Sci. 2004, 95, 448–453, doi:10.1111/j.1349-7006.2004.tb03230.x.
- Huang, Y.-L.; Wei, F.; Zhao, K.; Zhang, Y.; Wang, D.; Ya-Li, H. Isoliquiritigenin inhibits colorectal cancer cells HCT-116 growth by suppressing the PI3K/AKT pathway. Open Life Sci. 2017, 12, 300–307, doi:10.1515/biol-2017-0035.
- Sechet, E.; Telford, E.; Bonamy, C.; Sansonetti, P.J.; Sperandio, B. Natural molecules induce and synergize to boost expres-sion of the human antimicrobial peptide beta-defensin-3. Proc. Natl. Acad. Sci. USA 2018, 115, E9869––E9878.
- Chen, C.; Huang, S.; Chen, C.-L.; Su, S.-B.; Fang, D.-D. Isoliquiritigenin Inhibits Ovarian Cancer Metastasis by Reversing Epithelial-to-Mesenchymal Transition. Molecules 2019, 24, 3725, doi:10.3390/molecules24203725.
- Chen, H.-Y.; Huang, T.-C.; Shieh, T.-M.; Wu, C.-H.; Lin, L.-C.; Hsia, S.-M. Isoliquiritigenin Induces Autophagy and Inhibits Ovarian Cancer Cell Growth. Int. J. Mol. Sci. 2017, 18, 2025, doi:10.3390/ijms18102025.
- Mahalingam, S.; Gao, L.; Eisner, J.; Helferich, W.G.; Flaws, J.A. Effects of isoliquiritigenin on ovarian antral follicle growth and steroidogenesis. Reprod. Toxicol. 2016, 66, 107–114, doi:10.1016/j.reprotox.2016.10.004.
- Li, N.; Yang, L.; Deng, X.; Sun, Y. Effects of isoliquiritigenin on ovarian cancer cells. OncoTargets Ther. 2018, 11, 1633–1642, doi:10.2147/ott.s149295.
- Yuan, X.; Yu, B.; Wang, Y.; Jiang, J.; Liu, L.; Zhao, H.; Qi, W.; Zheng, Q. Involvement of endoplasmic reticulum stress in isoliquiritigenin-induced SKOV-3 cell apoptosis. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 191–199.
- Lee, J.-E.; Hong, E.-J.; Nam, H.-Y.; Hwang, M.; Kim, J.-H.; Han, B.-G.; Jeon, J.-P. Molecular signatures in response to Isoliqui-ritigenin in lymphoblastoid cell lines. Biochem. Biophys. Res. Commun. 2012, 427, 392–397, doi:10.1016/j.bbrc.2012.09.070.
- Li, D.; Wang, Z.; Chen, H.; Wang, J.; Zheng, Q.; Shang, J.; Li, J. Isoliquiritigenin induces monocytic differentiation of HL-60 cells. Free. Radic. Biol. Med. 2009, 46, 731–736, doi:10.1016/j.freeradbiomed.2008.11.011.
- Liu, Q.; Lv, H.; Wen, Z.; Ci, X.; Peng, L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-Like receptor protein 3 inflammasome and inhibits the NF-kappaB pathway in macrophages and in acute lung injury. Front. Immunol. 2017, 8, 1518.
- Traboulsi, H.; Cloutier, A.; Boyapelly, K.; Bonin, M.-A.; Marsault, É.; Cantin, A.M.; Richter, M.V. The Flavonoid Isoliquiriti-genin Reduces Lung Inflammation and Mouse Morbidity during Influenza Virus Infection. Antimicrob. Agents Chemother. 2015, 59, 6317–6327, doi:10.1128/aac.01098-15.
- Ho, W.; Zhou, Y. Combination of liquiritin, isoliquiritin and isoliquirigenin induce apoptotic cell death through upregu-lating p53 and p21 in the A549 non-small cell lung cancer cells. Oncol. Rep. 2013, 31, 298–304, doi:10.3892/or.2013.2849.
- Liu, B.; Yang, J.; Wen, Q.; Li, Y. Isoliquiritigenin, a flavonoid from licorice, relaxes guinea-pig tracheal smooth muscle in vitro and in vivo: Role of cGMP/PKG pathway. Eur. J. Pharmacol. 2008, 587, 257–266, doi:10.1016/j.ejphar.2008.03.015.
- Hsu, Y.-L.; Kuo, P.-L.; Chiang, L.-C.; Lin, C.-C. Isoliquiritigenin inhibits the proliferation and induces the apoptosis of hu-man non-small cell lung cancer a549 cells. Clin. Exp. Pharmacol. Physiol. 2004, 31, 414–418, doi:10.1111/j.1440-1681.2004.04016.x.
- Ii, T.; Satomi, Y.; Katoh, D.; Shimada, J.; Baba, M.; Okuyama, T.; Nishino, H.; Kitamura, N. Induction of cell cycle arrest and p21CIP1/WAF1 expression in human lung cancer cells by isoliquiritigenin. Cancer Lett. 2004, 207, 27–35, doi:10.1016/j.canlet.2003.10.023.
- Park, S.-J.; Song, H.-Y.; Youn, H.-S. Suppression of the TRIF-dependent signaling pathway of toll-like receptors by isoliqui-ritigenin in RAW264.7 macrophages. Mol. Cells 2009, 28, 365–368, doi:10.1007/s10059-009-0130-z.
- Lee, S.H.; Kim, J.Y.; Seo, G.S.; Kim, Y.-C.; Sohn, D.H. Isoliquiritigenin, from Dalbergia odorifera, up-regulates an-ti-inflammatory heme oxygenase-1 expression in RAW264.7 macrophages. Inflamm. Res. 2009, 58, 257–262, doi:10.1007/s00011-008-8183-6.
- Chen, H.; Zhang, B.; Yao, Y.; Chen, N.; Chen, X.; Tian, H.; Wang, Z.; Zheng, Q. NADPH Oxidase-Derived Reactive Oxygen Species Are Involved in the HL-60 Cell Monocytic Differentiation Induced by Isoliquiritigenin. Molecules 2012, 17, 13424–13438, doi:10.3390/molecules171113424.
- Chen, H.; Zhang, B.; Yuan, X.; Yao, Y.; Zhao, H.; Sun, X.; Zheng, Q. Isoliquiritigenin-induced effects on Nrf2 mediated anti-oxidant defence in the HL-60 cell monocytic differentiation. Cell Biol. Int. 2013, 37, 1215–1224, doi:10.1002/cbin.10156.
- Youns, M.; Fu, Y.-J.; Zu, Y.-G.; Kramer, A.; Konkimalla, V.B.; Radlwimmer, B.; Sültmann, H.; Efferth, T. Sensitivity and re-sistance towards isoliquiritigenin, doxorubicin and methotrexate in T cell acute lymphoblastic leukaemia cell lines by pharmacogenomics. Arch. Pharmacol. 2010, 382, 221–234, doi:10.1007/s00210-010-0541-6.
- Zu, Y.; Liu, X.; Fu, Y.-J.; Shi, X.; Wu, N.; Yao, L.; Efferth, T. Cytotoxic Activity of Isoliquiritigenin towards CCRF-CEM Leu-kemia Cells and its Effect on DNA Damage. Planta Med. 2009, 75, 1134–1140, doi:10.1055/s-0029-1185479.
- Yu, H.; Li, H.; Li, Y.; Li, M.; Chen, G. Effect of isoliquiritigenin for the treatment of atopic dermatitis-like skin lesions in mice. Arch. Dermatol. Res. 2017, 309, 805–813, doi:10.1007/s00403-017-1787-3.
- Xiang, S.; Chen, H.; Luo, X.-J.; An, B.; Wu, W.; Cao, S.; Ruan, S.; Wang, Z.; Weng, L.; Zhu, H.; et al. Isoliquiritigenin sup-presses human melanoma growth by targeting miR-301b/LRIG1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 184, doi:10.1186/s13046-018-0844-x.
- Chen, X.; Yang, M.; Hao, W.; Han, J.; Ma, J.; Wang, C.; Sun, S.; Zheng, Q. Differentiation-inducing and anti-proliferative ac-tivities of isoliquiritigenin and all-trans-retinoic acid on B16F0 melanoma cells: Mechanisms profiling by RNA-seq. Gene 2016, 592, 86–98, doi:10.1016/j.gene.2016.07.052.
- Chen, X.Y.; Li, D.F.; Han, J.C.; Wang, B.; Dong, Z.P.; Yu, L.N.; Pan, Z.H.; Qu, C.J.; Chen, Y.; Sun, S.G.; et al. Reprogramming induced by isoliquiritigenin diminishes melanoma cachexia through mTORC2-AKT-GSK3beta signaling. Oncotarget 2017, 8, 34565–34575.
- Chen, X.; Ren, H.-H.; Wang, D.; Chen, Y.; Qu, C.-J.; Pan, Z.-H.; Liu, X.; Hao, W.; Xu, W.-J.; Wang, K.; et al. Isoliquiritigenin Induces Mitochondrial Dysfunction and Apoptosis by Inhibiting mitoNEET in a Reactive Oxygen Species-Dependent Man-ner in A375 Human Melanoma Cells. Oxidative Med. Cell. Longev. 2019, 2019, 9817576, doi:10.1155/2019/9817576.
- Wang, Y.; Ma, J.; Yan, X.; Chen, X.; Si, L.; Liu, Y.; Han, J.; Hao, W.; Zheng, Q. Isoliquiritigenin Inhibits Proliferation and In-duces Apoptosis via Alleviating Hypoxia and Reducing Glycolysis in Mouse Melanoma B16F10 Cells. Recent Pat. An-ti-Cancer Drug Discov. 2016, 11, 215–227.
- Chen, X.; Zhang, B.; Yuan, X.; Yang, F.; Liu, J.; Zhao, H.; Liu, L.; Wang, Y.; Wang, Z.; Zheng, Q. Isoliquiritigenin-Induced Differentiation in Mouse Melanoma B16F0 Cell Line. Oxidative Med. Cell. Longev. 2012, 2012, 1–11, doi:10.1155/2012/534934.
- Chen, X.; Wu, Y.; Jiang, Y.; Zhou, Y.; Wang, Y.; Yao, Y.; Yi, C.; Gou, L.; Yang, J. Isoliquiritigenin inhibits the growth of mul-tiple myeloma via blocking IL-6 signaling. J. Mol. Med. 2012, 90, 1311–1319, doi:10.1007/s00109-012-0910-3.
- Lv, J.; Fu, Y.; Cao, Y.; Jiang, S.; Yang, Y.; Song, G.; Yun, C.; Gao, R. Isoliquiritigenin inhibits melanogenesis, melanocyte dendricity and melanosome transport by regulating ERK-mediated MITF degradation. Exp. Dermatol. 2020, 29, 149–157, doi:10.1111/exd.14066.
- Iwashita, K.; Kobori, M.; Yamaki, K.; Tsushida, T. Flavonoids Inhibit Cell Growth and Induce Apoptosis in B16 Melanoma 4A5 Cells. Biosci. Biotechnol. Biochem. 2000, 64, 1813–1820, doi:10.1271/bbb.64.1813.
- Huang, Y.; Liu, C.; Zeng, W.-C.; Xu, G.-Y.; Wu, J.-M.; Li, Z.-W.; Huang, X.-Y.; Lin, R.; Shi, X. Isoliquiritigenin inhibits the proliferation, migration and metastasis of Hep3B cells via suppressing cyclin D1 and PI3K/AKT pathway. Biosci. Rep. 2020, 40, 40, doi:10.1042/BSR20192727.
- Wang, J.R.; Luo, Y.H.; Piao, X.J.; Zhang, Y.; Feng, Y.C.; Li, J.Q.; Xu, W.T.; Zhang, Y.; Zhang, T.; Wang, S.N.; et al. Mechanisms underlying isoliquiritigenin-induced apoptosis and cell cycle arrest via ROS-mediated MAPK/STAT3/NF-kappaB pathways in human hepatocellular carcinoma cells. Drug Dev. Res. 2019, 80, 461–470.
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sci. 2005, 77, 279–292, doi:10.1016/j.lfs.2004.09.047.
- Hsu, Y.-L.; Kuo, P.-L.; Lin, L.-T.; Lin, C.-C. Isoliquiritigenin Inhibits Cell Proliferation and Induces Apoptosis in Human Hepatoma Cells. Planta Med. 2005, 71, 130–134, doi:10.1055/s-2005-837779.
- Jang, D.S.; Park, E.J.; Kang, Y.H.; Hawthorne, M.E.; Vigo, J.S.; Graham, J.G.; Cabieses, F.; Fong, H.H.; Mehta, R.G.; Pezzuto, J.M.; et al. Potential cncer chemopreventive flavonoids from the stems of Tephrosia toxicaria. J. Nat. Prod. 2003, 66, 1166–1170.
- Fang, S.-C.; Hsu, C.-L.; Lin, H.-T.; Yen, G.-C. Anticancer Effects of Flavonoid Derivatives Isolated from Millettia reticulata Benth in SK-Hep-1 Human Hepatocellular Carcinoma Cells. J. Agric. Food Chem. 2010, 58, 814–820, doi:10.1021/jf903216r.
- Zhang, B.; Lai, Y.; Li, Y.; Shu, N.; Wang, Z.; Wang, Y.; Li, Y.; Chen, Z. Antineoplastic activity of isoliquiritigenin, a chalcone compound, in androgen-independent human prostate cancer cells linked to G2/M cell cycle arrest and cell apoptosis. Eur. J. Pharmacol. 2018, 821, 57–67, doi:10.1016/j.ejphar.2017.12.053.
- Lee, Y.M.; Lim, D.Y.; Choi, H.J.; Jung, J.I.; Chung, W.-Y.; Park, J.H.Y. Induction of Cell Cycle Arrest in Prostate Cancer Cells by the Dietary Compound Isoliquiritigenin. J. Med. Food 2009, 12, 8–14, doi:10.1089/jmf.2008.0039.
- Kwon, G.T.; Cho, H.J.; Chung, W.-Y.; Park, K.-K.; Moon, A.; Park, J.H.Y. Isoliquiritigenin inhibits migration and invasion of prostate cancer cells: Possible mediation by decreased JNK/AP-1 signaling. J. Nutr. Biochem. 2009, 20, 663–676, doi:10.1016/j.jnutbio.2008.06.005.
- Jung, J.I.; Chung, E.; Seon, M.R.; Shin, H.-K.; Kim, E.J.; Lim, S.S.; Chung, W.-Y.; Park, K.-K.; Park, J.H.Y. Isoliquiritigenin (ISL) inhibits ErbB3 signaling in prostate cancer cells. BioFactors 2006, 28, 159–168, doi:10.1002/biof.5520280302.
- Jung, J.I.; Lim, S.S.; Choi, H.J.; Shin, H.-K.; Kim, E.J.; Chung, W.-Y.; Park, K.-K.; Park, J.H.Y. Isoliquiritigenin induces apop-tosis by depolarizing mitochondrial membranes in prostate cancer cells. J. Nutr. Biochem. 2006, 17, 689–696, doi:10.1016/j.jnutbio.2005.11.006.
- Kanazawa, M.; Satomi, Y.; Mizutani, Y.; Ukimura, O.; Kawauchi, A.; Sakai, T.; Baba, M.; Okuyama, T.; Nishino, H.; Miki, T. Isoliquiritigenin Inhibits the Growth of Prostate Cancer. Eur. Urol. 2003, 43, 580–586, doi:10.1016/s0302-2838(03)00090-3.
- Hirchaud, F.; Hermetet, F.; Ablise, M.; Fauconnet, S.; Vuitton, D.A.; Prétet, J.-L.; Mougin, C. Isoliquiritigenin Induces Caspase-Dependent Apoptosis via Downregulation of HPV16 E6 Expression in Cervical Cancer Ca Ski Cells. Planta Med. 2013, 79, 1628–1635, doi:10.1055/s-0033-1350956.
- Yuan, X.; Zhang, B.; Gan, L.; Wang, Z.H.; Yu, B.C.; Liu, L.L.; Zheng, Q.; Wang, Z.P. Involvement of the mitochondri-on-dependent and the endoplasmic reticulum stress-signaling pathways in isoliquiritigenin-induced apoptosis of HeLa cell. Biomed. Environ. Sci. 2013, 26, 268–276.
- Hsu, Y.-L.; Chia, C.-C.; Chen, P.-J.; Huang, S.-E.; Huang, S.-C.; Kuo, P.-L. Shallot and licorice constituent isoliquiritigenin arrests cell cycle progression and induces apoptosis through the induction of ATM/p53 and initiation of the mitochondrial system in human cervical carcinoma HeLa cells. Mol. Nutr. Food Res. 2009, 53, 826–835, doi:10.1002/mnfr.200800288.
- Zhang, X.; Wang, S.; Sun, W.; Wei, C. Isoliquiritigenin inhibits proliferation and metastasis of MKN28 gastric cancer cells by suppressing the PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 18, 3429–3436, doi:10.3892/mmr.2018.9318.
- Lee, H.H.; Lee, S.; Shin, Y.-S.; Cho, M.; Kang, H.J.; Cho, H. Anti-Cancer Effect of Quercetin in Xenograft Models with EBV-Associated Human Gastric Carcinoma. Molecules 2016, 21, 1286, doi:10.3390/molecules21101286.
- Ma, J.; Fu, N.-Y.; Pang, D.-B.; Wu, W.-Y.; Xu, A.-L. Apoptosis Induced by Isoliquiritigenin in Human Gastric Cancer MGC-803 Cells. Planta Med. 2001, 67, 754–757, doi:10.1055/s-2001-18361.
- Kim, D.-C.; Ramachandran, S.; Baek, S.-H.; Kwon, S.-H.; Kwon, K.-Y.; Cha, S.-D.; Bae, I.; Cho, C.-H. Induction of Growth In-hibition and Apoptosis in Human Uterine Leiomyoma Cells by Isoliquiritigenin. Reprod. Sci. 2008, 15, 552–558, doi:10.1177/1933719107312681.
- Chen, J.; Liu, C.; Yang, Q.-Q.; Ma, R.-B.; Ke, Y.; Dong, F.; Wu, X.-E. Isoliquiritigenin Suppresses Osteosarcoma U2OS Cell Proliferation and Invasion by Regulating the PI3K/Akt Signalling Pathway. Chemotherapy 2018, 63, 155–161, doi:10.1159/000490151.
- Li, C.; Zhou, X.; Sun, C.; Liu, X.; Shi, X.; Wu, S. Isoliquiritigenin inhibits the proliferation, apoptosis and migration of oste-osarcoma cells. Oncol. Rep. 2019, 41, 2502–2510, doi:10.3892/or.2019.6998.
- Alshangiti, A.M.; Togher, K.L.; Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. The dietary flavonoid isoliquiritigenin is a potent cytotoxin for human neuroblastoma cells. Neuronal Signal. 2019, 3, 201, doi:10.1042/ns20180201.
- Zhao, S.; Chang, H.; Ma, P.; Gao, G.; Jin, C.; Zhao, X.; Zhou, W.; Jin, B. Inhibitory effect of DNA topoisomerase inhibitor isoliquiritigenin on the growth of glioma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12577–12582.
- Yang, H.-H.; Zhang, C.; Lai, S.-H.; Zeng, C.-C.; Liu, Y.; Wang, X.-Z. Isoliquiritigenin Induces Cytotoxicity in PC-12 Cells In Vitro. Appl. Biochem. Biotechnol. 2017, 183, 1173–1190, doi:10.1007/s12010-017-2491-7.
- Si, L.; Yang, X.; Yanming, W.; Wang, Y.; Zheng, Q. Isoliquiritigenin induces apoptosis of human bladder cancer T24 cells via a cyclin-dependent kinase-independent mechanism. Oncol. Lett. 2017, 14, 241–249, doi:10.3892/ol.2017.6159.

