Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Richard Y Zhao | -- | 3011 | 2024-03-18 16:12:13 | | | |
| 2 | Lindsay Dong | Meta information modification | 3011 | 2024-03-19 03:03:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, J.; Hom, K.; Zhang, C.; Nasr, M.; Gerzanich, V.; Zhang, Y.; Tang, Q.; Xue, F.; Simard, J.M.; Zhao, R.Y. SARS-CoV-2 ORF3a Protein as Therapeutic Target against COVID-19. Encyclopedia. Available online: https://encyclopedia.pub/entry/56386 (accessed on 25 June 2026).
Zhang J, Hom K, Zhang C, Nasr M, Gerzanich V, Zhang Y, et al. SARS-CoV-2 ORF3a Protein as Therapeutic Target against COVID-19. Encyclopedia. Available at: https://encyclopedia.pub/entry/56386. Accessed June 25, 2026.
Zhang, Jiantao, Kellie Hom, Chenyu Zhang, Mohamed Nasr, Volodymyr Gerzanich, Yanjin Zhang, Qiyi Tang, Fengtian Xue, J. Marc Simard, Richard Y. Zhao. "SARS-CoV-2 ORF3a Protein as Therapeutic Target against COVID-19" Encyclopedia, https://encyclopedia.pub/entry/56386 (accessed June 25, 2026).
Zhang, J., Hom, K., Zhang, C., Nasr, M., Gerzanich, V., Zhang, Y., Tang, Q., Xue, F., Simard, J.M., & Zhao, R.Y. (2024, March 18). SARS-CoV-2 ORF3a Protein as Therapeutic Target against COVID-19. In Encyclopedia. https://encyclopedia.pub/entry/56386
Zhang, Jiantao, et al. "SARS-CoV-2 ORF3a Protein as Therapeutic Target against COVID-19." Encyclopedia. Web. 18 March, 2024.
Copy Citation
The ORF3a (open reading frame 3a) protein found in SARS-CoV-2, represents a promising target for antiviral treatment due to its multifaceted role in viral pathogenesis, cytokine storms, disease severity, and mortality. ORF3a contributes significantly to viral pathogenesis by facilitating viral assembly and release, essential processes in the viral life cycle, while also suppressing the body’s antiviral responses, thus aiding viral replication. ORF3a also has been implicated in triggering excessive inflammation, characterized by NF-κB-mediated cytokine production, ultimately leading to apoptotic cell death and tissue damage in the lungs, kidneys, and the central nervous system.
SARS-CoV-2
COVID-19
ORF3a
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2 infection was unprecedented in its rapid rate of spread, continuous emergence of new viral variants, and the high number of mortality cases and post-COVID complications. While vaccines have generally demonstrated efficacy in preventing SARS-CoV-2 infection or mitigating the severity of COVID-19, their effectiveness has often been compromised by the emergence of new viral variants and a decline in efficacy over time [1]. Promising antiviral drugs such as Paxlovid and molnupiravir have shown potential in treating COVID-19 and alleviating long COVID symptoms. However, challenges such as viral rebound and drug resistance have been observed [2][3]. Moreover, despite the pandemic showing signs of waning, a substantial number of infections persist worldwide, with the continued emergence of new viral variants. Given the relatively high mutable nature of coronaviruses (CoVs), there is an ongoing need to develop new antiviral drugs.
The development of antiviral drugs holds pivotal importance in the battle against COVID-19. Antiviral drugs play a critical role in various facets of disease management. For instance, remdesivir, an antiviral drug designed to impede SARS-CoV-2 replication, has demonstrated effectiveness in reducing the severity and duration of illness in COVID-19 patients [4]. Additionally, such medications diminish the viral load in infected individuals, thereby reducing the risk of transmitting the virus to others and aiding in community control. Antiviral drug development is essential in tackling emerging viral variants with diverse resistance profiles against existing treatments, necessitating ongoing research and development. Antiviral drugs can complement vaccination efforts, providing an added layer of protection in scenarios where vaccines are not universally available or effective due to the emergence of new viral variants.
2. SARS-CoV-2 ORF3a Protein as a Promising Antiviral Target
To address the need for discovering novel antiviral targets against COVID-19, a comprehensive genome-wide functional analysis of SARS-CoV-2 proteins was conducted, utilizing a fission yeast cell-based system [5][6]. The selection criteria focused on identifying a viral therapeutic target crucial for viral pathogenesis associated with COVID-19 and exhibiting a quantifiable endpoint suitable for high-throughput screening (HTS), such as cell proliferation or cell death. Utilizing a fission yeast gene expression vector, a total of 29 SARS-CoV-2 viral proteins [7], covering the entire genome of the SARS-CoV-2 reference strain USA-WA1/2020 (GenBank#: MN985325) [8], were cloned [6][9][10]. Each viral gene was placed under an inducible nmt1 promoter [5][10][11], ensuring a specific measurement of the effects caused by the viral protein. Of the 29 SARS-CoV-2 viral proteins studied, the ORF3a (open reading frame 3a) protein uniquely met all the predefined selection criteria, emerging as a potential therapeutic target [12][13].
Increasing evidence underlines the clinical relevance of the ORF3a protein, as both SARS-CoV-1 and SARS-CoV-2-infected patients exhibited high levels of anti-ORF3a antibodies, and sera from recovering COVID-19 patients displayed considerable IgG and IgA reactivity specifically against ORF3a [14][15][16][17]. Notably, the IgG response to ORF3a was linked to severe COVID-19 across different age groups [18]. Robust SARS-CoV-2-specific T-cell responses targeting ORF3a were also observed in asymptomatic individuals or COVID-19 convalescent patients [19][20][21].
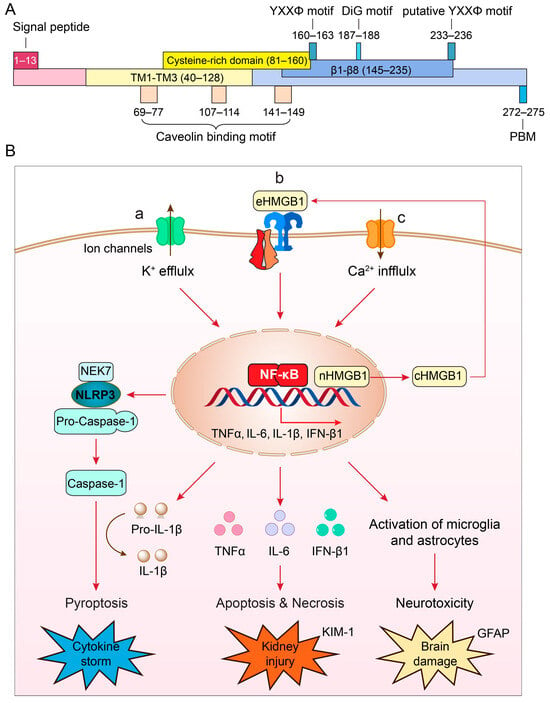
3. Structure and Unique Features of ORF3a Protein
ORF3a is the largest accessory protein of SARS-CoV-2, displaying numerous functions that are not yet fully characterized (Figure 1A). Comparison of ORF3a protein sequences between SARS-CoV-1 and SARS-CoV-2 suggests that these two proteins share 73% sequence homology, with SARS-CoV-1 ORF3a also lacking one amino acid at position 241 (E241) [13][22]. Consequently, the SARS-CoV-2 ORF3a protein sequence is unique, and its three-dimensional (3D) tertiary structure is also novel, as it bears no resemblance to other known proteins [22][23].
Typically found as a homodimer or tetramer on the cell membrane [22], each monomer of ORF3a comprises 275 amino acids (aa), with a calculated molecular mass of 31 kD (Figure 1A). It harbors three hydrophobic transmembrane domains (TM1–TM3; aa40–128), anchoring it within the membrane. Both the N- and C-termini face the cytoplasm, housing distinct functional domains responsible for varied functionalities, including intracellular transport, ion channel formation and/or modulation, cytopathic effects, virus release, and virus production [13][23][24][25].
Figure 1. (A) Structure and unique features of SARS-CoV-2 ORF3a protein (adapted from [13][22]). The nucleotide sequence of ORF3a is 825 bp in length and encodes a 31 kD protein of 275 amino acids. It shows an extracellular N-terminal signal peptide (aa1–13), 3 transmembrane (TM) domains (aa40–128) that are across the cellular membrane and 8 antiparallel β-sheets (β1–β8, aa145–235) at the cytoplasmic C-terminus. Other functional motifs include 3 caveolin-binding motifs (aa69–77, aa107–114 and aa141–149) [26]; a YXXΦ motif (aa160–163) and a diG motif (aa177–178) [25][27][28], and a PBM (aa272–275) [13][29]. A putative YXXΦ motif (aa233–236) is also indicated [30]. (B) Cellular pathways of ORF3a-mediated effects that lead to the NLRP3 inflammasome activation and cytokine storm (a) [31][32][33], NF-κB-mediated cytokine production, apoptosis and necrosis leading to kidney injury (b) [34], and disruption of autophagy-lysosomal pathway by ORF3a that causes reactive microglia and astrocyte activation leading to neuroinflammation and brain damage (c) [35]. Elevation of kidney injury molecule 1 (KIM-1) is a specific biomarker for kidney injury [36]; elevation of glial fibrillary acidic protein (GFAP) is a well-accepted biomarker for brain damage [37][38].
The subgenomic ORF3a sequence is transcribed within the viral replication-transcription complex (RTC) of the double-membrane vesicles (DMVs) [13][39]. DMVs, formed by the rearrangement of the endoplasmic reticulum (ER), are also impacted by ORF3a, as it disrupts ER organization when ORF3a is overproduced [40].
ORF3a serves as a viroporin [13][22][23][41][42][43]–a viral transmembrane protein demonstrating ion channel properties within cell membranes [44]. Viroporins typically exhibit ion channel activities that can impact cell membrane permeability, Ca2+ homeostasis, and membrane remodeling. Their primary functions often involve virion morphogenesis, viral entry, replication, and the release (egress) of viruses [44]. Specifically, SARS-CoV-2 ORF3a forms a nonselective calcium (Ca2+)-permeable cation channel within a liposome system [22]. This channel allows the passage of large cations including NMDG+ and YO-PRO-1, resembling other Ca2+-permeable channels such as Trpv1, Trpa1, and P2x7 [22].
The ORF3a protein exhibits several well-conserved functional motifs (Figure 1A), allowing us to hypothesize about its potential functions. Notably, it contains a TNF receptor-associated factor 3 (TRAF3)-binding motif spanning aa36–40 in both SARS-CoV-1 and SARS-CoV-2 [31][32][45][46].
ORF3a harbors three putative caveolin-binding motifs located at aa69–77, aa107–114, and aa141–149 [24][26]. These motifs potentially regulate the intracellular trafficking of ORF3a [24][26]. Caveolins, membrane proteins involved in caveolae formation (small invaginations of the plasma membrane), might influence endocytosis during viral entry.
Early mutational analyses have pinpointed specific functional domains within the ORF3a protein, notably the YXXΦ motif (aa160–163) and the diG motif (aa187–188) situated in the cytoplasmic region. These domains play pivotal roles in directing the intracellular transport of ORF3a from the ER to lysosomes [25][27][28]. The YXXΦ motif, where Y signifies tyrosine, X represents any amino acid, and Φ denotes a bulky hydrophobic residue such as leucine or phenylalanine, is a recognized tyrosine-based sorting motif crucial for mediating the intracellular trafficking of proteins within the cell.
The ORF3a protein possesses a highly conserved PDZ-binding motif (PBM) at its C-terminal end (SVPL; aa272–275) [13][29]. Given that there are more than 400 host cellular PDZ-containing proteins, the presence of the PBM in ORF3a suggests its potential to interact with a broad spectrum of host cellular functions [47][48]. It is a common tactic of pathogenic viruses, including SARS-CoV-2, to target cellular PDZ proteins [49].
4. Role of ORF3a in Viral Production
ORF3a plays a pivotal role in multiple processes contributing to viral pathogenesis, particularly in viral assembly and release, which are crucial steps in the viral life cycle. SARS-CoV-2 ORF3a, but not SARS-CoV-1, facilitates viral release via the lysosomal exocytosis pathway [50]. ORF3a promotes this process by facilitating the trafficking of lysosomes to the plasma membrane and the fusion of exocytosis-related SNARE vesicle fusion proteins. This is achieved by aiding in the lysosomal targeting of the BORC-ARL8b complex [50]. Notably, SARS-CoV-2 ORF3a-mediated lysosomal exocytosis necessitates the activity of the Ca2+ channel TRPML3 [50]. Cells producing SARS-CoV-2 ORF3a demonstrated elevated cytosolic Ca2+ concentration compared to control cells, and TRPML3 knockdown impedes ORF3a-mediated lysosomal exocytosis [50]. Additionally, viruses such as mouse hepatitis virus (MHV), which lack ORF3a or SARS-CoV-1, exhibit reduced efficiency in releasing viral particles [50][51][52].
ORF3a serves as a potent virulence factor contributing significantly to viral pathogenesis. Deletion of ORF3a from the SARS-CoV-2 genome results in a considerable reduction in viral production within human cells or transgenic mice [53][54][55]. Although ORF3a is not the sole contributor to viral production, combined deletion of ORF3a with other accessory proteins, ORF6, 7, and 8 (Δ3678), significantly weakens the virus, making it a potential candidate for a live attenuated virus vaccine [56]. Notably, while the Δ678 virus (without ORF3a deletion) had a mild effect, the Δ3678 virus exhibited a 3-log reduction in virus yield compared to the wildtype virus, underscoring the substantial contribution of ORF3a to the decrease in viral production [55][56]. Consistent evidence from studies, such as a CoronaVac vaccine investigation using inactivated virus, demonstrates a significant increase in antibody titers for ORF3a at two weeks post-COVID-19 diagnosis [57].
5. Role of ORF3a in Viral Pathogenesis: Induction of Cytokine Storm and COVID-19 Severity
The onset of a cytokine storm is a primary contributor to COVID-19-related morbidity and mortality [31][42][58][59][60]. These storms involve an excessive release of pro-inflammatory cytokines and chemokines, leading to hyperinflammation, acute respiratory distress syndrome, and multi-organ failure, often resulting in fatality. Overactive or dysregulated NLRP3 inflammasome activation in SARS-CoV-2-infected patients is linked to triggering cytokine storms, causing tissue damage and severe organ failure in individuals with critical COVID-19 [61][62].
The ORF3a protein has been implicated in the observed cytokine storms in severe CoV infections (Figure 1B-a) [33][63]. Specifically, ORF3a induces a pro-inflammatory immune response in infected cells, activating the NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome [31][32][33][42]. This multimeric protein complex triggers the secretion of pro-inflammatory cytokines [61][64][65], resulting in a hyper-inflammatory response. Such data indicate the substantial contribution of ORF3a to the severity and fatality of COVID-19 [31][42][58][59][60].
The exact molecular mechanism underpinning NLRP3 inflammasome activation by ORF3a remains elusive, but several models have been proposed: (1) ion channel activity, in which ORF3a exhibits ion channel activity disrupting cellular ion homeostasis, particularly calcium (Ca2+) and potassium (K+), leading to intracellular ion imbalances that trigger cellular stress responses, mitochondrial dysfunction, and eventual NLRP3 inflammasome activation [33][42]; (2) mitochondrial dysfunction, in which ORF3a’s interaction with mitochondria might induce damage or dysfunction, resulting in the release of mitochondrial reactive oxygen species (ROS), which could activate the NLRP3 inflammasome via the NIM811-sensitive mitochondrial-permeability-pore (mtPTP) [33]; and (3) direct NLRP3 inflammasome activation, in which ORF3a might directly activate the NLRP3 inflammasome [13][33], acting via HMGB1 (high-mobility group box 1), a downstream effector of the NLRP3 inflammasome to induce caspase-1-mediated pyroptosis (Figure 1B-b) [62][66][67]. HMGB1 is a ubiquitous protein released by microglia or macrophages upon NLRP3 inflammasome activation, and it promotes TLR4- and RAGE (receptor for advanced glycation end products) receptor-mediated pro-inflammatory cytokine production [68][69]. Elevated serum HMGB1 levels in COVID-19 patients correlate with poor prognosis [70][71]. HMGB1 inhibitors, like glycyrrhizin, reduce ORF3a-induced HMGB1 release and production of pro-inflammatory cytokines, potentially inhibiting SARS-CoV-2 replication [67].
6. Roles of ORF3a in Kidney Injury, Neuroinflammation, and Long-COVID Complications
Among millions of COVID-related deaths in the U.S. and worldwide, between a quarter and half of patients died with kidney complications such as acute kidney injury (AKI) [72][73][74]. AKI is a sudden decline of the normal kidney function that ranges from minor kidney malfunction to complete failure that could result in death. Patients with acute respiratory failure often also have AKI and have the worst overall prognosis [75][76].
Although AKI is a major contributor to COVID-19-related death [74], the underlying cause of death in SARS-CoV-2-related AKI remains elusive. Besides the lungs, SARS-CoV-2 also infects the kidney via the ACE2 and other receptors such as CD209L/L-SIGN, CD209/DC-SIGN [77] and BSG/CD147 [78] in renal proximal tubular epithelial cells (RPTEC), where it supports kidney integrity and function [79][80]. Both SARS-CoV-2 RNA and proteins have been detected in SARS-CoV-2-infected kidneys [81][82], and viral particles recovered from the kidney were able to infect nonhuman primate RPTEC [83]. Histopathological examinations, including ours, biopsy or postmortem kidney tissues, showed that acute injury in RPTEC is most common in COVID-19 patients with AKI [72][84][85][86]. Those with acute tubular injury are associated with oxidative stress and inflammation-mediated cytokine release such as TNFα and IL-6 [72][84][85][86], which results in apoptosis and necrosis [87][88][89].
Mechanistically, ORF3a induces renal tubule injuries by activation of NF-κB and STAT3 (activator of transcription 3) signaling [34]. ORF3a promotes STAT3 activation through interaction with a ubiquitin E3 ligase TRIM59, by which it dissociates the phosphatase TC-PTP from binding to STAT3, presumably by protein degradation, and hence it inhibits the dephosphorylation of STAT3, leading to persistent STAT3 activation [34].
SARS-CoV-2 may also infects the central nerve system (CNS) [90], leading to various neurological complications associated with COVID-19, both short- and long-term [91][92][93]. A national cohort study on patients with COVID-19 showed that COVID-19-associated neurologic complications link to a higher risk of disability and death [94]. Specifically, the virus affects brain glial astrocytes and induces neuroinflammation and neurotoxicity [92][93][95].
7. Discovery and Development of Inhibitor Drugs Targeting ORF3a Protein
7.1. Testing Well-Established Druggable Cellular Targets of Key ORF3a Regulators with FDA-Approved Drugs and Small Molecule Inhibitors (SMIs)
ORF3a-mediated signaling pathways involve key cellular regulators such as NLRP3, NF-κB, and related cytokines (TNFα and IL-6), all well-known druggable cellular targets relevant to COVID-19 [12][13]. A promising approach is to repurpose FDA-approved drugs that target these regulators against ORF3a-mediated pathways. For instance, ORF3a activates NLRP3, triggering cytokine storms linked to tissue damage and organ failure in severe COVID-19 cases (Figure 1B-a) [42][96][97][98]. Several compounds have shown potential as anti-NLRP3 inhibitors in preclinical studies or early clinical trials. MCC950 (CRID3) emerged as a notable NLRP3 inhibitor, demonstrating efficacy in various inflammatory disease models [99].
The FDA-approved drug glibenclamide (glyburide) targets the Sur1-Trpm4 ion channel to inhibit NLRP3 inflammasome activation in microglia, effectively mitigating neuroinflammation [100]. Given that ORF3a, acting as a viroporin, triggers NLRP3 inflammasome activation and microglial responses in the brain [35], exploring whether ORF3a initiates NLRP3 inflammasome activation via the Sur1-Trpm4 channel is of great interest. Investigating whether glibenclamide can specifically impede ORF3a-induced NLRP3 inflammasome activation presents a compelling avenue for potential interventions against neuroinflammatory responses linked to SARS-CoV-2 infections. Such studies could offer valuable insights into managing the neurological complications associated with COVID-19.
NF-κB stands as another significant target to counteract the effects of ORF3a (Figure 1B-b), being a pivotal regulator in pro-inflammatory signaling pathways, where NLRP3 inflammasome activation is facilitated through NF-κB [42]. Auranofin, an FDA-approved NF-κB inhibitor, has displayed promising outcomes in impeding SARS-CoV-2 infection and associated inflammatory damages [101], warranting further examination against ORF3a-mediated responses.
Considering that ORF3a stimulates NF-κB to induce cytokine production such as TNFα and IL-6 [12][45][102], and that elevation of TNFα and IL-6 are two strong and independent survival predictors of patients with COVID-19 [103][104], targeting these cytokines is also appealing. TNFα and IL-6 serve as well-established druggable cellular targets with several FDA-approved anti-TNFα or IL-6 drugs either suggested for COVID-19 treatment or in various clinical trials [105][106].
HMGB1 emerges as another promising druggable cellular target to counteract the effects of ORF3a, given its role in promoting HMGB1 secretion by ORF3a, which subsequently activates NF-κB and NLRP3 inflammasome (Figure 1B-c) [13][33].
7.2. Structure-Based Design of ORF3a Inhibitors
The cryo-EM structure of ORF3a is established [22] yet identifying specific target sites for precise inhibition remains elusive. Initial molecular docking studies using in silico models have highlighted potential interactions between ORF3a and various macroheterocyclic compounds, including chlorin e6 and cationic porphyrin such as TmPyP4 [107]. Subsequent analyses using fluorescence and UV-vis spectroscopy confirmed the interaction of chlorin e6 and porphyrin with ORF3a protein [107]. An intriguing aspect is that TmPyP4 is known for intercalating into and stabilizing G-quadruplexes (G4), which are present in DNA and RNA sequences containing guanine blocks [108].
7.3. Cell-Based High-Throughput Drug Screening
Cell-based high-throughput drug screening stands as a promising strategy for discovering and developing anti-ORF3a inhibitors, offering several advantages: (1) it enables comprehensive searches across diverse drug libraries, enhancing the potential to uncover a broader spectrum of inhibitors; (2) the screening process is target-specific, focusing exclusively on ORF3a production; (3) unlike structure-based drug design, this method is functionally driven, allowing for the identification of various inhibitors, including novel types such as allosteric inhibitors that act on ORF3a regardless of their binding to the ORF3a protein; and (4) the process automatically filters out cytotoxic compounds from HTS drug screenings.
Apart from employing mammalian cell based HTS, yeasts like fission yeast (Schizosaccharomyces pombe) have also been utilized in antiviral drug screenings via HTS [5][109][110]. Fission yeast presents distinct advantages compared to mammalian cell systems. For instance, it is easily maintainable and exhibits rapid growth, making it particularly well-suited for extensive drug screening.
The use of yeast in HTS does not necessitate mammalian complexities but requires a functionally conserved output to indicate the presence of ORF3a, which need not necessarily correlate with viral or host cell biology. As an illustration, HIV-1 protease (PR)-induced cell death in fission yeast has served as a metric for HTS, distinct from its PR enzymatic activity, used to screen for HIV-1 PR inhibitors (PIs) [110][111][112].
The key of using fission yeast cell-based system to study ORF3a is that the measured ORF3a effect on cellular processes must be highly conserved between yeast and human cells, particularly in fundamental cellular events such as cell proliferation and cell death [5][113][114].
As ORF3a represents a viral protein, any potential ORF3a inhibitors identified through HTS require further validation by testing within the context of viral infection. Various standardized protocols have been developed and utilized for screening small molecule inhibitors against SARS-CoV-2 infection [8][115].
References
- Alcendor, D.J.; Matthews-Juarez, P.; Smoot, D.; Hildreth, J.E.K.; Lamar, K.; Tabatabai, M.; Wilus, D.; Juarez, P.D. Breakthrough COVID-19 Infections in the US: Implications for Prolonging the Pandemic. Vaccines 2022, 10, 755.
- Rubin, R. From Positive to Negative to Positive Again-The Mystery of Why COVID-19 Rebounds in Some Patients Who Take Paxlovid. JAMA 2022, 327, 2380–2382.
- Service, R.F. Bad news for Paxlovid? Resistance may be coming. Science 2022, 377, 138–139.
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826.
- Zhao, R.Y. Yeast for virus research. Microb. Cell 2017, 4, 311–330.
- Li, G.; Zhao, R.Y. Molecular Cloning and Characterization of Small Viral Genome in Fission Yeast. Methods Mol. Biol. 2018, 1721, 47–61.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266–1273.
- Zhao, Y.; Elder, R.T.; Chen, M.; Cao, J. Fission yeast expression vectors adapted for positive identification of gene insertion and green fluorescent protein fusion. Biotechniques 1998, 25, 438–440, 442, 444.
- Nkeze, J.; Li, L.; Benko, Z.; Li, G.; Zhao, R.Y. Molecular characterization of HIV-1 genome in fission yeast Schizosaccharomyces pombe. Cell Biosci. 2015, 5, 47.
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385.
- Zhang, J.; Li, Q.; Cruz Cosme, R.S.; Gerzanich, V.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Genome-Wide Characterization of SARS-CoV-2 Cytopathogenic Proteins in the Search of Antiviral Targets. mBio 2021, 13, e0016922.
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567.
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.H.; Chu, D.K.W.; Mok, C.K.P.; Tsang, O.T.Y.; Yeung, Y.C.; Perera, R.; Poon, L.L.M.; et al. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS-CoV-2 infection. Nat. Immunol. 2020, 21, 1293–1301.
- Camerini, D.; Randall, A.Z.; Trappl-Kimmons, K.; Oberai, A.; Hung, C.; Edgar, J.; Shandling, A.; Huynh, V.; Teng, A.A.; Hermanson, G.; et al. Mapping SARS-CoV-2 Antibody Epitopes in COVID-19 Patients with a Multi-Coronavirus Protein Microarray. Microbiol. Spectr. 2021, 9, e0141621.
- Liu, I.J.; Hsueh, P.R.; Lin, C.T.; Chiu, C.Y.; Kao, C.L.; Liao, M.Y.; Wu, H.C. Disease-specific B Cell epitopes for serum antibodies from patients with severe acute respiratory syndrome (SARS) and serologic detection of SARS antibodies by epitope-based peptide antigens. J. Infect. Dis. 2004, 190, 797–809.
- Zhong, X.; Guo, Z.; Yang, H.; Peng, L.; Xie, Y.; Wong, T.Y.; Lai, S.T.; Guo, Z. Amino terminus of the SARS coronavirus protein 3a elicits strong, potentially protective humoral responses in infected patients. J. Gen. Virol. 2006, 87, 369–373.
- Zhu, A.; Liu, M.; Li, Y.; Lei, Q.; Wu, Q.; Lin, M.; Lai, D.; Lu, L.; Yu, S.; Guo, S.; et al. Age- and Severity-Associated Humoral Immunity Response in COVID-19 Patients: A Cohort Study from Wuhan, China. J. Clin. Med. 2022, 11, 5974.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e1415.
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728.
- Samandari, T.; Ongalo, J.B.; McCarthy, K.D.; Biegon, R.K.; Madiega, P.A.; Mithika, A.; Orinda, J.; Mboya, G.M.; Mwaura, P.; Anzala, O.; et al. Prevalence and functional profile of SARS-CoV-2 T cells in asymptomatic Kenyan adults. J. Clin. Investig. 2023, 133, 13.
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582.
- McClenaghan, C.; Hanson, A.; Lee, S.J.; Nichols, C.G. Coronavirus Proteins as Ion Channels: Current and Potential Research. Front. Immunol. 2020, 11, 573339.
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20.
- Cruz-Cosme, R.; Zhang, J.; Liu, D.; Mahase, V.; Sallapalli, B.T.; Chang, P.; Zhang, Y.; Teng, S.; Zhao, R.Y.; Tang, Q. A novel diG motif in ORF3a protein of SARS-CoV-2 for intracellular transport. Front. Cell Dev. Biol. 2022, 10, 1011221.
- Padhan, K.; Tanwar, C.; Hussain, A.; Hui, P.Y.; Lee, M.Y.; Cheung, C.Y.; Peiris, J.S.M.; Jameel, S. Severe acute respiratory syndrome coronavirus Orf3a protein interacts with caveolin. J. Gen. Virol. 2007, 88, 3067–3077.
- Tan, Y.J.; Teng, E.; Shen, S.; Tan, T.H.; Goh, P.Y.; Fielding, B.C.; Ooi, E.E.; Tan, H.C.; Lim, S.G.; Hong, W. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J. Virol. 2004, 78, 6723–6734.
- Minakshi, R.; Padhan, K. The YXXPhi motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol. J. 2014, 11, 75.
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826.
- Kakkanas, A.; Karamichali, E.; Koufogeorgou, E.I.; Kotsakis, S.D.; Georgopoulou, U.; Foka, P. Targeting the YXXPhi Motifs of the SARS Coronaviruses 1 and 2 ORF3a Peptides by In Silico Analysis to Predict Novel Virus-Host Interactions. Biomolecules 2022, 12, 1052.
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877.
- Jin, D.Y.; Zheng, B.J.; Tang, H.M.V. Mechanism of inflammasome activation by SARS coronavirus 3a protein: Abridged secondary publication. Hong Kong Med. J. 2021, 27 (Suppl. S2), 33–35.
- Guarnieri, J.W.; Angelin, A.; Murdock, D.G.; Schaefer, P.; Portluri, P.; Lie, T.; Huang, J.; Wallace, D.C. SARS-CoV-2 viroporins activate the NLRP3-inflammasome by the mitochondrial permeability transition pore. Front. Immunol. 2023, 14, 1064293.
- Cai, H.; Chen, Y.; Feng, Y.; Asadi, M.; Kaufman, L.; Lee, K.; Kehrer, T.; Miorin, L.; Garcia-Sastre, A.; Gusella, G.L.; et al. SARS-CoV-2 viral protein ORF3A injures renal tubules by interacting with TRIM59 to induce STAT3 activation. Mol. Ther. 2023, 31, 774–787.
- Zhu, H.; Byrnes, C.; Lee, Y.T.; Tuymetova, G.; Duffy, H.B.D.; Bakir, J.Y.; Pettit, S.N.; Angina, J.; Springer, D.A.; Allende, M.L.; et al. SARS-CoV-2 ORF3a expression in brain disrupts the autophagy-lysosomal pathway, impairs sphingolipid homeostasis, and drives neuropathogenesis. FASEB J. 2023, 37, e22919.
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487.
- Schiff, L.; Hadker, N.; Weiser, S.; Rausch, C. A literature review of the feasibility of glial fibrillary acidic protein as a biomarker for stroke and traumatic brain injury. Mol. Diagn. Ther. 2012, 16, 79–92.
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136.
- Lee, Y.B.; Jung, M.; Kim, J.; Charles, A.; Christ, W.; Kang, J.; Kang, M.G.; Kwak, C.; Klingstrom, J.; Smed-Sorensen, A.; et al. Super-resolution proximity labeling reveals anti-viral protein network and its structural changes against SARS-CoV-2 viral proteins. Cell Rep. 2023, 42, 112835.
- Breitinger, U.; Farag, N.S.; Sticht, H.; Breitinger, H.G. Viroporins: Structure, function, and their role in the life cycle of SARS-CoV-2. Int. J. Biochem. Cell Biol. 2022, 145, 106185.
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22.
- Fam, M.S.; Sedky, C.A.; Turky, N.O.; Breitinger, H.G.; Breitinger, U. Channel activity of SARS-CoV-2 viroporin ORF3a inhibited by adamantanes and phenolic plant metabolites. Sci. Rep. 2023, 13, 5328.
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574.
- Zhang, J.; Cruz-Cosme, R.; Zhang, C.; Liu, D.; Tang, Q.; Zhao, R.Y. Endoplasmic reticulum-associated SARS-CoV-2 ORF3a elicits heightened cytopathic effects despite robust ER-associated degradation. mBio 2023, e0303023.
- Busscher, B.M.; Befekadu, H.B.; Liu, Z.; Xiao, T.S. SARS-CoV-2 ORF3a-Mediated NF-kappaB Activation Is Not Dependent on TRAF-Binding Sequence. Viruses 2023, 15, 2229.
- Caillet-Saguy, C.; Durbesson, F.; Rezelj, V.V.; Gogl, G.; Tran, Q.D.; Twizere, J.C.; Vignuzzi, M.; Vincentelli, R.; Wolff, N. Host PDZ-containing proteins targeted by SARS-CoV-2. FEBS J. 2021, 288, 5148–5162.
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018, 9, 10–1128.
- Rice, A.P.; Kimata, J.T. SARS-CoV-2 likely targets cellular PDZ proteins: A common tactic of pathogenic viruses. Future Virol. 2021, 16, 375–377.
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell 2021, 56, 3250–3263.e5.
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545.
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514.
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005, 79, 14909–14922.
- Akerstrom, S.; Mirazimi, A.; Tan, Y.J. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antivir. Res. 2007, 73, 219–227.
- Silvas, J.A.; Vasquez, D.M.; Park, J.G.; Chiem, K.; Allue-Guardia, A.; Garcia-Vilanova, A.; Platt, R.N.; Miorin, L.; Kehrer, T.; Cupic, A.; et al. Contribution of SARS-CoV-2 Accessory Proteins to Viral Pathogenicity in K18 Human ACE2 Transgenic Mice. J. Virol. 2021, 95, e0040221.
- Liu, Y.; Zhang, X.; Liu, J.; Xia, H.; Zou, J.; Muruato, A.E.; Periasamy, S.; Kurhade, C.; Plante, J.A.; Bopp, N.E.; et al. A live-attenuated SARS-CoV-2 vaccine candidate with accessory protein deletions. Nat. Commun. 2022, 13, 4337.
- Duarte, L.F.; Vazquez, Y.; Diethelm-Varela, B.; Pavez, V.; Berrios-Rojas, R.; Mendez, C.; Riedel, C.A.; White, J.A.; Kalergis, A.M.; Bueno, S.M.; et al. Differential Severe Acute Respiratory Syndrome Coronavirus 2-Specific Humoral Response in Inactivated Virus-Vaccinated, Convalescent, and Breakthrough-Infected Subjects. J. Infect. Dis. 2023, 228, 857–867.
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell Mol. Immunol. 2021, 18, 1305–1307.
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729.
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256.
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518.
- Van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front. Immunol. 2020, 11, 1580.
- Bertoni, A.; Penco, F.; Mollica, H.; Bocca, P.; Prigione, I.; Corcione, A.; Cangelosi, D.; Schena, F.; Del Zotto, G.; Amaro, A.; et al. Spontaneous NLRP3 inflammasome-driven IL1-beta secretion is induced in severe COVID-19 patients and responds to anakinra treatment. J. Allergy Clin. Immunol. 2022, 150, 796–805.
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128.
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484.
- Gowda, P.; Patrick, S.; Joshi, S.D.; Kumawat, R.K.; Sen, E. Glycyrrhizin prevents SARS-CoV-2 S1 and Orf3a induced high mobility group box 1 (HMGB1) release and inhibits viral replication. Cytokine 2021, 142, 155496.
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628.
- Peng, T.; Du, S.Y.; Son, M.; Diamond, B. HIF-1alpha is a negative regulator of interferon regulatory factors: Implications for interferon production by hypoxic monocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2106017118.
- Bolay, H.; Karadas, O.; Ozturk, B.; Sonkaya, R.; Tasdelen, B.; Bulut, T.D.S.; Gulbahar, O.; Ozge, A.; Baykan, B. HMGB1, NLRP3, IL-6 and ACE2 levels are elevated in COVID-19 with headache: A window to the infection-related headache mechanism. J. Headache Pain 2021, 22, 94.
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 992–994.
- Legrand, M.; Bell, S.; Forni, L.; Joannidis, M.; Koyner, J.L.; Liu, K.; Cantaluppi, V. Pathophysiology of COVID-19-associated acute kidney injury. Nat. Rev. Nephrol. 2021, 17, 751–764.
- May, R.M.; Cassol, C.; Hannoudi, A.; Larsen, C.P.; Lerma, E.V.; Haun, R.S.; Braga, J.R.; Hassen, S.I.; Wilson, J.; VanBeek, C.; et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. 2021, 100, 1303–1315.
- Bowe, B.; Cai, M.; Xie, Y.; Gibson, A.K.; Maddukuri, G.; Al-Aly, Z. Acute Kidney Injury in a National Cohort of Hospitalized US Veterans with COVID-19. Clin. J. Am. Soc. Nephrol. 2020, 16, 14–25.
- Chan, L.; Chaudhary, K.; Saha, A.; Chauhan, K.; Vaid, A.; Zhao, S.; Paranjpe, I.; Somani, S.; Richter, F.; Miotto, R.; et al. AKI in Hospitalized Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 151–160.
- Geri, G.; Darmon, M.; Zafrani, L.; Fartoukh, M.; Voiriot, G.; Le Marec, J.; Nemlaghi, S.; Vieillard-Baron, A.; Azoulay, E. Acute kidney injury in SARS-CoV2-related pneumonia ICU patients: A retrospective multicenter study. Ann. Intensive Care 2021, 11, 86.
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165.
- Kalejaiye, T.D.; Bhattacharya, R.; Burt, M.A.; Travieso, T.; Okafor, A.E.; Mou, X.; Blasi, M.; Musah, S. SARS-CoV-2 Employ BSG/CD147 and ACE2 Receptors to Directly Infect Human Induced Pluripotent Stem Cell-Derived Kidney Podocytes. Front. Cell Dev. Biol. 2022, 10, 855340.
- Khan, S.; Chen, L.; Yang, C.R.; Raghuram, V.; Khundmiri, S.J.; Knepper, M.A. Does SARS-CoV-2 Infect the Kidney? J. Am. Soc. Nephrol. 2020, 31, 2746–2748.
- Armaly, Z.; Kinaneh, S.; Skorecki, K. Renal Manifestations of COVID-19: Physiology and Pathophysiology. J. Clin. Med. 2021, 10, 1216.
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592.
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227.
- Braun, F.; Lutgehetmann, M.; Pfefferle, S.; Wong, M.N.; Carsten, A.; Lindenmeyer, M.T.; Norz, D.; Heinrich, F.; Meissner, K.; Wichmann, D.; et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 2020, 396, 597–598.
- Papadimitriou, J.C.; Drachenberg, C.B.; Kleiner, D.; Choudhri, N.; Haririan, A.; Cebotaru, V. Tubular Epithelial and Peritubular Capillary Endothelial Injury in COVID-19 AKI. Kidney Int. Rep. 2021, 6, 518–525.
- Alexander, M.P.; Mangalaparthi, K.K.; Madugundu, A.K.; Moyer, A.M.; Adam, B.A.; Mengel, M.; Singh, S.; Herrmann, S.M.; Rule, A.D.; Cheek, E.H.; et al. Acute Kidney Injury in Severe COVID-19 Has Similarities to Sepsis-Associated Kidney Injury: A Multi-Omics Study. Mayo Clin. Proc. 2021, 96, 2561–2575.
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506.
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200.
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144.
- Guo, L.P.; Liu, S.X.; Yang, Q.; Liu, H.Y.; Xu, L.L.; Hao, Y.H.; Zhang, X.Q. Effect of Thymoquinone on Acute Kidney Injury Induced by Sepsis in BALB/c Mice. Biomed. Res. Int. 2020, 2020, 1594726.
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175.
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269.
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.; Vendramini, P.H.; Valenca, A.G.F.; Brandao-Teles, C.; Zuccoli, G.D.S.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119.
- Dedoni, S.; Avdoshina, V.; Camoglio, C.; Siddi, C.; Fratta, W.; Scherma, M.; Fadda, P. K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research. Molecules 2022, 27, 4142.
- Xu, E.; Xie, Y.; Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022, 28, 2406–2415.
- Marshall, M. How COVID-19 can damage the brain. Nature 2020, 585, 342–343.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50.
- Bakhshi, S.; Shamsi, S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: Latest evidence and therapeutic outcomes. Int. Immunopharmacol. 2022, 106, 108595.
- He, Y.; Chang, Y.; Peng, Y.; Zhu, J.; Liu, K.; Chen, J.; Wu, Y.; Ji, Z.; Lin, Z.; Wang, S.; et al. Glibenclamide Directly Prevents Neuroinflammation by Targeting SUR1-TRPM4-Mediated NLRP3 Inflammasome Activation in Microglia. Mol. Neurobiol. 2022, 59, 6590–6607.
- Laplantine, E.; Chable-Bessia, C.; Oudin, A.; Swain, J.; Soria, A.; Merida, P.; Gourdelier, M.; Mestiri, S.; Besseghe, I.; Bremaud, E.; et al. The FDA-approved drug Auranofin has a dual inhibitory effect on SARS-CoV-2 entry and NF-kappaB signaling. iScience 2022, 25, 105066.
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464.
- Sayah, W.; Berkane, I.; Guermache, I.; Sabri, M.; Lakhal, F.Z.; Yasmine Rahali, S.; Djidjeli, A.; Lamara Mahammed, L.; Merah, F.; Belaid, B.; et al. Interleukin-6, procalcitonin and neutrophil-to-lymphocyte ratio: Potential immune-inflammatory parameters to identify severe and fatal forms of COVID-19. Cytokine 2021, 141, 155428.
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643.
- Izadi, Z.; Brenner, E.J.; Mahil, S.K.; Dand, N.; Yiu, Z.Z.N.; Yates, M.; Ungaro, R.C.; Zhang, X.; Agrawal, M.; Colombel, J.F.; et al. Association Between Tumor Necrosis Factor Inhibitors and the Risk of Hospitalization or Death Among Patients with Immune-Mediated Inflammatory Disease and COVID-19. JAMA Netw. Open 2021, 4, e2129639.
- Lonze, B.E.; Spiegler, P.; Wesson, R.N.; Alachkar, N.; Petkova, E.; Weldon, E.P.; Dieter, R.A.; Li, Y.; Quinn, M.; Mattoo, A.; et al. A Randomized Double-Blinded Placebo Controlled Trial of Clazakizumab for the Treatment of COVID-19 Pneumonia with Hyperinflammation. Crit. Care Med. 2022, 50, 1348–1359.
- Lebedeva, N.S.; Gubarev, Y.A.; Mamardashvili, G.M.; Zaitceva, S.V.; Zdanovich, S.A.; Malyasova, A.S.; Romanenko, J.V.; Koifman, M.O.; Koifman, O.I. Theoretical and experimental study of interaction of macroheterocyclic compounds with ORF3a of SARS-CoV-2. Sci. Rep. 2021, 11, 19481.
- Arora, A.; Maiti, S. Differential biophysical behavior of human telomeric RNA and DNA quadruplex. J. Phys. Chem. B 2009, 113, 10515–10520.
- Benko, Z.; Elder, R.T.; Liang, D.; Zhao, R.R. Fission yeast as a HTS platform for molecular probes of HIV-1 Vpr-induced cell death. Int. J. High Throughput Screen. 2010, 1, 151–162.
- Benko, Z.; Zhang, J.; Zhao, R.Y. Development of A Fission Yeast Cell-Based Platform for High Throughput Screening of HIV-1 Protease Inhibitors. Curr. HIV Res. 2019, 17, 429–440.
- Benko, Z.; Elder, R.T.; Li, G.; Liang, D.; Zhao, R.Y. HIV-1 Protease in the Fission Yeast Schizosaccharomyces pombe. PLoS ONE 2016, 11, e0151286.
- Benko, Z.; Liang, D.; Li, G.; Elder, R.T.; Sarkar, A.; Takayama, J.; Ghosh, A.K.; Zhao, R.Y. A fission yeast cell-based system for multidrug resistant HIV-1 proteases. Cell Biosci. 2017, 7, 5.
- Zhao, Y.; Elder, R.T. Yeast perspectives on HIV-1 Vpr. Front. Biosci. 2000, 5, 905–916.
- Andreola, M.L.; Litvak, S. Yeast and the AIDS virus: The odd couple. J. Biomed. Biotechnol. 2012, 2012, 549020.
- Chu, H.; Chan, J.F.; Yuen, T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.; Tsang, J.O.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
826
Revisions:
2 times
(View History)
Update Date:
19 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No