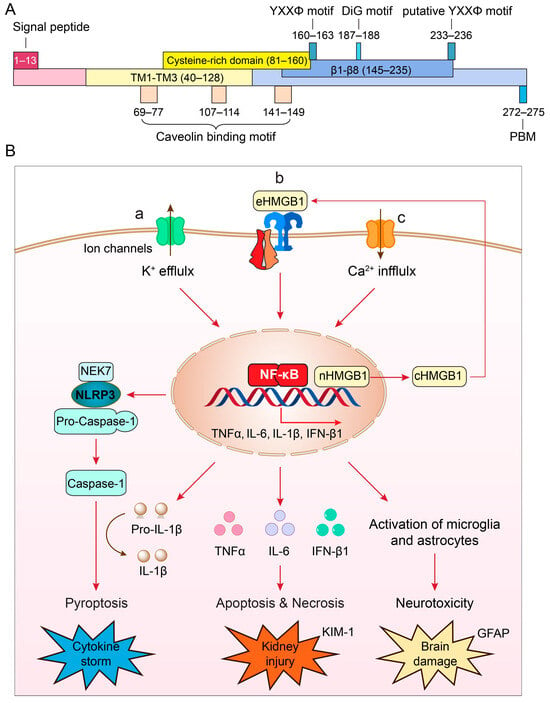
ORF3a serves as a potent virulence factor contributing significantly to viral pathogenesis. Deletion of ORF3a from the SARS-CoV-2 genome results in a considerable reduction in viral production within human cells or transgenic mice [
]. Although ORF3a is not the sole contributor to viral production, combined deletion of ORF3a with other accessory proteins, ORF6, 7, and 8 (Δ3678), significantly weakens the virus, making it a potential candidate for a live attenuated virus vaccine [
]. Notably, while the Δ678 virus (without ORF3a deletion) had a mild effect, the Δ3678 virus exhibited a 3-log reduction in virus yield compared to the wildtype virus, underscoring the substantial contribution of ORF3a to the decrease in viral production [
]. Consistent evidence from studies, such as a CoronaVac vaccine investigation using inactivated virus, demonstrates a significant increase in antibody titers for ORF3a at two weeks post-COVID-19 diagnosis [
The onset of a cytokine storm is a primary contributor to COVID-19-related morbidity and mortality [
37,
51,
74,
75,
76]. These storms involve an excessive release of pro-inflammatory cytokines and chemokines, leading to hyperinflammation, acute respiratory distress syndrome, and multi-organ failure, often resulting in fatality. Overactive or dysregulated NLRP3 inflammasome activation in SARS-CoV-2-infected patients is linked to triggering cytokine storms, causing tissue damage and severe organ failure in individuals with critical COVID-19 [
77,
78].
The ORF3a protein has been implicated in the observed cytokine storms in severe CoV infections (
Figure 1B-a) [
39,
79]. Specifically, ORF3a induces a pro-inflammatory immune response in infected cells, activating the NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome [
37,
38,
39,
51]. This multimeric protein complex triggers the secretion of pro-inflammatory cytokines [
77,
80,
81], resulting in a hyper-inflammatory response. Such data indicate the substantial contribution of ORF3a to the severity and fatality of COVID-19 [
37,
51,
74,
75,
76].
The exact molecular mechanism underpinning NLRP3 inflammasome activation by ORF3a remains elusive, but several models have been proposed: (1) ion channel activity, in which ORF3a exhibits ion channel activity disrupting cellular ion homeostasis, particularly calcium (Ca
2+) and potassium (K
+), leading to intracellular ion imbalances that trigger cellular stress responses, mitochondrial dysfunction, and eventual NLRP3 inflammasome activation [
39,
51]; (2) mitochondrial dysfunction, in which ORF3a’s interaction with mitochondria might induce damage or dysfunction, resulting in the release of mitochondrial reactive oxygen species (ROS), which could activate the NLRP3 inflammasome via the NIM811-sensitive mitochondrial-permeability-pore (mtPTP) [
39]; and (3) direct NLRP3 inflammasome activation, in which ORF3a might directly activate the NLRP3 inflammasome [
18,
39], acting via HMGB1 (high-mobility group box 1), a downstream effector of the NLRP3 inflammasome to induce caspase-1-mediated pyroptosis (
Figure 1B-b) [
78,
88,
89]. HMGB1 is a ubiquitous protein released by microglia or macrophages upon NLRP3 inflammasome activation, and it promotes TLR4- and RAGE (receptor for advanced glycation end products) receptor-mediated pro-inflammatory cytokine production [
90,
91]. Elevated serum HMGB1 levels in COVID-19 patients correlate with poor prognosis [
92,
93]. HMGB1 inhibitors, like glycyrrhizin, reduce ORF3a-induced HMGB1 release and production of pro-inflammatory cytokines, potentially inhibiting SARS-CoV-2 replication [
89].
6. Roles of ORF3a in Kidney Injury, Neuroinflammation, and Long-COVID Complications
Among millions of COVID-related deaths in the U.S. and worldwide, between a quarter and half of patients died with kidney complications such as acute kidney injury (AKI) [
94,
95,
96]. AKI is a sudden decline of the normal kidney function that ranges from minor kidney malfunction to complete failure that could result in death. Patients with acute respiratory failure often also have AKI and have the worst overall prognosis [
97,
98].
Although AKI is a major contributor to COVID-19-related death [
96], the underlying cause of death in SARS-CoV-2-related AKI remains elusive. Besides the lungs, SARS-CoV-2 also infects the kidney via the ACE2 and other receptors such as CD209L/L-SIGN, CD209/DC-SIGN [
101] and BSG/CD147 [
102] in renal proximal tubular epithelial cells (RPTEC), where it supports kidney integrity and function [
103,
104]. Both SARS-CoV-2 RNA and proteins have been detected in SARS-CoV-2-infected kidneys [
99,
105], and viral particles recovered from the kidney were able to infect nonhuman primate RPTEC [
106]. Histopathological examinations, including ours, biopsy or postmortem kidney tissues, showed that acute injury in RPTEC is most common in COVID-19 patients with AKI [
94,
107,
108,
109]. Those with acute tubular injury are associated with oxidative stress and inflammation-mediated cytokine release such as TNFα and IL-6 [
94,
107,
108,
109], which results in apoptosis and necrosis [
110,
111,
112].
Mechanistically, ORF3a induces renal tubule injuries by activation of NF-κB and STAT3 (activator of transcription 3) signaling [
40]. ORF3a promotes STAT3 activation through interaction with a ubiquitin E3 ligase TRIM59, by which it dissociates the phosphatase TC-PTP from binding to STAT3, presumably by protein degradation, and hence it inhibits the dephosphorylation of STAT3, leading to persistent STAT3 activation [
40].
SARS-CoV-2 may also infects the central nerve system (CNS) [
114], leading to various neurological complications associated with COVID-19, both short- and long-term [
115,
116,
117]. A national cohort study on patients with COVID-19 showed that COVID-19-associated neurologic complications link to a higher risk of disability and death [
118]. Specifically, the virus affects brain glial astrocytes and induces neuroinflammation and neurotoxicity [
116,
117,
119].
7. Discovery and Development of Inhibitor Drugs Targeting ORF3a Protein
7.1. Testing Well-Established Druggable Cellular Targets of Key ORF3a Regulators with FDA-Approved Drugs and Small Molecule Inhibitors (SMIs)
ORF3a-mediated signaling pathways involve key cellular regulators such as NLRP3, NF-κB, and related cytokines (TNFα and IL-6), all well-known druggable cellular targets relevant to COVID-19 [
17,
18]. A promising approach is to repurpose FDA-approved drugs that target these regulators against ORF3a-mediated pathways. For instance, ORF3a activates NLRP3, triggering cytokine storms linked to tissue damage and organ failure in severe COVID-19 cases (
Figure 1B-a) [
51,
82,
83,
84]. Several compounds have shown potential as anti-NLRP3 inhibitors in preclinical studies or early clinical trials. MCC950 (CRID3) emerged as a notable NLRP3 inhibitor, demonstrating efficacy in various inflammatory disease models [
122].
The FDA-approved drug glibenclamide (glyburide) targets the Sur1-Trpm4 ion channel to inhibit NLRP3 inflammasome activation in microglia, effectively mitigating neuroinflammation [
124]. Given that ORF3a, acting as a viroporin, triggers NLRP3 inflammasome activation and microglial responses in the brain [
41], exploring whether ORF3a initiates NLRP3 inflammasome activation via the Sur1-Trpm4 channel is of great interest. Investigating whether glibenclamide can specifically impede ORF3a-induced NLRP3 inflammasome activation presents a compelling avenue for potential interventions against neuroinflammatory responses linked to SARS-CoV-2 infections. Such studies could offer valuable insights into managing the neurological complications associated with COVID-19.
NF-
κB stands as another significant target to counteract the effects of ORF3a (
Figure 1B-b), being a pivotal regulator in pro-inflammatory signaling pathways, where NLRP3 inflammasome activation is facilitated through NF-κB [
51]. Auranofin, an FDA-approved NF-κB inhibitor, has displayed promising outcomes in impeding SARS-CoV-2 infection and associated inflammatory damages [
125], warranting further examination against ORF3a-mediated responses.
Considering that ORF3a stimulates NF-κB to induce cytokine production such as TNFα and IL-6 [
17,
47,
126], and that elevation of TNFα and IL-6 are two strong and independent survival predictors of patients with COVID-19 [
127,
128], targeting these cytokines is also appealing. TNFα and IL-6 serve as well-established druggable cellular targets with several FDA-approved anti-TNFα or IL-6 drugs either suggested for COVID-19 treatment or in various clinical trials [
129,
130].
HMGB1 emerges as another promising druggable cellular target to counteract the effects of ORF3a, given its role in promoting HMGB1 secretion by ORF3a, which subsequently activates NF-κB and NLRP3 inflammasome (
Figure 1B-c) [
18,
39].
7.2. Structure-Based Design of ORF3a Inhibitors
The cryo-EM structure of ORF3a is established [
28] yet identifying specific target sites for precise inhibition remains elusive. Initial molecular docking studies using in silico models have highlighted potential interactions between ORF3a and various macroheterocyclic compounds, including chlorin e6 and cationic porphyrin such as TmPyP4 [
133]. Subsequent analyses using fluorescence and UV-vis spectroscopy confirmed the interaction of chlorin e6 and porphyrin with ORF3a protein [
133]. An intriguing aspect is that TmPyP4 is known for intercalating into and stabilizing G-quadruplexes (G4), which are present in DNA and RNA sequences containing guanine blocks [
134].
7.3. Cell-Based High-Throughput Drug Screening
Cell-based high-throughput drug screening stands as a promising strategy for discovering and developing anti-ORF3a inhibitors, offering several advantages: (1) it enables comprehensive searches across diverse drug libraries, enhancing the potential to uncover a broader spectrum of inhibitors; (2) the screening process is target-specific, focusing exclusively on ORF3a production; (3) unlike structure-based drug design, this method is functionally driven, allowing for the identification of various inhibitors, including novel types such as allosteric inhibitors that act on ORF3a regardless of their binding to the ORF3a protein; and (4) the process automatically filters out cytotoxic compounds from HTS drug screenings.
Apart from employing mammalian cell based HTS, yeasts like fission yeast (
Schizosaccharomyces pombe) have also been utilized in antiviral drug screenings via HTS [
10,
137,
138]. Fission yeast presents distinct advantages compared to mammalian cell systems. For instance, it is easily maintainable and exhibits rapid growth, making it particularly well-suited for extensive drug screening.
The use of yeast in HTS does not necessitate mammalian complexities but requires a functionally conserved output to indicate the presence of ORF3a, which need not necessarily correlate with viral or host cell biology. As an illustration, HIV-1 protease (PR)-induced cell death in fission yeast has served as a metric for HTS, distinct from its PR enzymatic activity, used to screen for HIV-1 PR inhibitors (PIs) [
138,
139,
140].
The key of using fission yeast cell-based system to study ORF3a is that the measured ORF3a effect on cellular processes must be highly conserved between yeast and human cells, particularly in fundamental cellular events such as cell proliferation and cell death [
10,
144,
145].
As ORF3a represents a viral protein, any potential ORF3a inhibitors identified through HTS require further validation by testing within the context of viral infection. Various standardized protocols have been developed and utilized for screening small molecule inhibitors against SARS-CoV-2 infection [
13,
149].