Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alessandra Baracca | -- | 4341 | 2024-02-27 17:38:35 | | | |
| 2 | Jason Zhu | Meta information modification | 4341 | 2024-02-28 07:13:34 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Del Dotto, V.; Musiani, F.; Baracca, A.; Solaini, G. Biochemical Dysfunctions Related to MT-ATP6/MT-ATP8 Pathogenic Variants. Encyclopedia. Available online: https://encyclopedia.pub/entry/55565 (accessed on 28 July 2026).
Del Dotto V, Musiani F, Baracca A, Solaini G. Biochemical Dysfunctions Related to MT-ATP6/MT-ATP8 Pathogenic Variants. Encyclopedia. Available at: https://encyclopedia.pub/entry/55565. Accessed July 28, 2026.
Del Dotto, Valentina, Francesco Musiani, Alessandra Baracca, Giancarlo Solaini. "Biochemical Dysfunctions Related to MT-ATP6/MT-ATP8 Pathogenic Variants" Encyclopedia, https://encyclopedia.pub/entry/55565 (accessed July 28, 2026).
Del Dotto, V., Musiani, F., Baracca, A., & Solaini, G. (2024, February 27). Biochemical Dysfunctions Related to MT-ATP6/MT-ATP8 Pathogenic Variants. In Encyclopedia. https://encyclopedia.pub/entry/55565
Del Dotto, Valentina, et al. "Biochemical Dysfunctions Related to MT-ATP6/MT-ATP8 Pathogenic Variants." Encyclopedia. Web. 27 February, 2024.
Copy Citation
Mitochondrial ATP synthase (Complex V) catalyzes the last step of oxidative phosphorylation and provides most of the energy (ATP) required by human cells. The mitochondrial genes MT-ATP6 and MT-ATP8 encode two subunits of the multi-subunit Complex V. Since the discovery of the first MT-ATP6 variant in the year 1990 as the cause of Neuropathy, Ataxia, and Retinitis Pigmentosa (NARP) syndrome, a large and continuously increasing number of inborn variants in the MT-ATP6 and MT-ATP8 genes have been identified as pathogenic. Variants in these genes correlate with various clinical phenotypes, which include several neurodegenerative and multisystemic disorders.
mitochondria
ATP synthase
ATP6
ATP8
mt-DNA
1. Introduction
The mitochondrial genome presents some specific features, including maternal inheritance and heteroplasmy. Heteroplasmy is a condition in which at least two different mitochondrial genomes are present within the same cell. Pathogenic variants in the mt-DNA are highly recessive and usually coexist with the wild-type mt-DNA molecules. Therefore, the clinical manifestation of mt-DNA variants mainly depends on both their severity and the mutational load (heteroplasmy) of the tissues [1][2].
Over the last two decades, a large number of studies using different patient’s specimens and other cellular paradigms have led to in-depth investigations of the biochemical and cellular alterations caused by MT-ATP6 and MT-ATP8 variants, which are summarized in Table 1.
As shown in Table 1, the cellular dysfunctions observed for a given variant can be highly variable, and one element that contributes to this characteristic is heteroplasmy.
Consequently, as with other mt-DNA-associated diseases, a specific feature is the threshold of the percentage of mutant genome (or percentage of heteroplasmy) that must be exceeded to detect a biochemical alteration. As reported below, this defect may also depend on the variant, haplogroup, cell type, and tissue type.
Table 1. Biochemical and cellular parameters in patient tissues and cell models carrying MT-ATP6 and MT-ATP8 pathogenic variants. Abbreviation: heteroplasmy (H), oxygen consumption rate (OCR), mitochondrial membrane potential (MMP), reactive oxygen species (ROS), induced pluripotent stem cell (iPSC) and neural progenitor cells (NPCs), Normal (N), Decreased (D), Increased (I), Affected (A).
| Genetic Variant/Subunit AA Change | Tissue or Cell Models H (%) |
Biochemical and Cellular Parameters | ||||
|---|---|---|---|---|---|---|
| CV ATP Synthesis |
CV ATP Hydrolysis |
OCR | CV Assembly/Stability | Other Mitochondrial and Cellular Readouts |
||
| m.8382C>T ATP8: p.T6I |
Muscle (100%) [3] |
(D) | CI activity (D) | |||
| Fibroblasts (100%) [3] |
(N) | (N) | CIV activity (D) | |||
| m.8403T>C ATP8: p.I13T |
Fibroblasts (100%) |
(N) [3] | (N) [3] | Depolarized plasma membrane and ROS (I) [4]; CIV activity (D) [3] |
||
| Yeast (100%) [5] |
(D) | (N) | Growth in stress conditions (D); Mitochondrial membrane potential (N) |
|||
| m.8424T>C ATP8: p.I20P |
Muscle (100%) [3] |
(D) | CI, CII, CIII, and CIV activities (D) | |||
| Fibroblasts (100%) [3] |
(D) | (N) | CI activity and growth in galactose media (D) | |||
| Cybrids (100%) [3] |
(D) | (D) | CI and CIV activities (D); Lactate production (I) |
|||
| m.8528T>C ATP8: p.W55R ATP6: p.M1T |
Fibroblast (93%) [6] |
(D) | ||||
| Heart muscle (90%) [7] | (A) | CV subunit levels and CI activity (D); ATP6 and ATP8 protein levels (D) |
||||
| m.8529G>A ATP8: p.W55X ATP6: p.M1M |
Muscle (>90%) [8] |
(D) | (D) | (A) | CI-CIV activities (N) | |
| Fibroblast (>90%) [8] |
(D) | CI-CIV activities (N) | ||||
| Cybrids (100%) |
(D) [8][9] | (D) [9] | (A) [8][9] | Growth in galactose media (D); ATP6 and ATP8 protein levels (D); Complexes II, III, and IV levels (D) [9] |
||
| m.8561C>G ATP8: p.P66A ATP6: p.P12R |
Myoblasts (99%) [10] |
(A) | Total ATP level (D); ROS (N); ATP6 and ATP8 protein levels (N) |
|||
| m.8561C>T ATP8: p.P66L ATP6: p.P12S |
Muscle (99%) [11] | (D) | (A) | |||
| m.8611insC ATP6: p.L29PfsX36 |
Muscle (60%) [12] |
(D) | (A) | |||
| Fibroblasts (80%) [12] |
(D) | (A) | ATP6 protein level (D); Mitochondrial cristae structure and dynamics (A) |
|||
| m.8618insT ATP6: p.T33HfsX32 |
Muscle (65–85%) |
(A) [13][14] | ATP6 protein level (D) [13] | |||
| Fibroblasts (45%) [14] |
(D) | (A) | ROS (I); Mitochondrial network morphology (N) |
|||
| m.8648G>A ATP6: p.R41Q |
Fibroblast (100%) [3] |
(N) | (N) | |||
| Cybrids (100%) [3] |
(N) | (N) | ||||
| m.8782G>A ATP6: p.G86X |
Fibroblasts (12–27%) [14] |
(D) | (A) | ROS (I); Mitochondrial morphology (N) |
||
| m.8806C>G ATP6: p.P94A |
Muscle (100%) [3] |
(N) | CI-CIV activities (D) | |||
| m.8839G>C ATP6: p.A105P |
Cybrids (100%) [15] |
(N) | Growth in galactose media (D); Mt-DNA copy number (I); OXPHOS protein levels (I); Mitochondrial membrane potential (D); CI-CIV activities (N) |
|||
| m.8843T>C ATP6: p.I106T |
Yeast (100%) [16] |
(N) | (N) | (N) | Mitochondrial membrane potential (N) | |
| m.8851T>C ATP6: p.W109R |
Yeast (100%) [17] |
(D) | (D) | (D) | (N) | Growth in stress conditions (D); Mitochondrial cristae structure (A); CIII and CIV super-complexes (D) |
| m.8909T>C ATP6: p.F128S |
Yeast (100%) [18] |
(D) | (D) | (A) | ||
| m.8932C>T ATP6: p.P136S |
Yeast (100%) [19] |
(D) | (D) | (A) | ATP6 protein level (D) | |
| m.8946A>C ATP6: p.M140I |
Fibroblasts (100%) [3] |
(N) | (N) | CI activity (D) | ||
| m.8950G>A ATP6: p.V142I |
Lymphocytes [20] | (D) | ||||
| Yeast (100%) [16] | (D) | (D) | (N) | Sensitivity of growth to oligomycin (I); Mitochondrial membrane potential (N) |
||
| m.8969G>A ATP6: p.S148N |
Muscle (100%) [3] | (N) | CI activity (D) | |||
| Yeast (100%) |
(D) [21][22] | (D) [21] | (D) [21][22] | (A) [21] | Growth in stress conditions (D) [21][22] | |
| Cybrids (19–98%) [21] |
(D) | Mitochondrial cristae structure (A); ROS (I) |
||||
| Fibroblasts (100%) [23] |
(D) | |||||
| m.8975T>C ATP6: p.L150P |
Muscle [3] | (D) | CI activity (D) | |||
| Fibroblasts (100%) [3] |
(D) | (N) | CI activity (D); Growth in galactose media (D) |
|||
| Cybrids (100%) [3] |
(N) | (N) | CI and CIV activities (D); Growth in galactose media (D); Lactate production (I) |
|||
| m.8989G>C ATP6: p.A155P |
Muscle (92%) [24] |
(D) | Mitochondrial ultrastructure (N) | |||
| m.8993T>G ATP6: p.L156R |
Yeast (100%) [25] |
(D) | (D) | (D) | (A) | Growth in stress conditions (D); CIV level (D) |
| Platelets (80–93%) |
(D) [26][27] | (N) [26][27] | CV ATP-driven proton flow (N) [26] | |||
| Lymphocytes (80–100%) |
(D) [28][29][30][31] | (D) [28] | (D) [29] | ROS and mitochondrial membrane potential (I) [31]; CV proton flow (D) [30][32]; Oligomycin sensitivity of CV proton flow (I) [32] |
||
| Muscle (76%) [33] |
(A) | |||||
| Fibroblasts (70–100%) |
(D) [3][34][35][36][37][38][39] | (D) [35][38][40]; (N) [3][34][36] | (D) [41][42]; (N) [34] |
(N) [36][37] | Mitochondrial membrane potential (I) [36][39]; Mitochondrial morphology (A) [39][42]; ROS (I) [39][43]; Antioxidant enzymes (A) [39]; Oligomycin sensitivity of CV (I) [34]; Growth in galactose media (D) [3][35][38][40]; Mitochondrial calcium uptake (D) [39]; Glycolytic capacity (D) [42]; CI and CIV activities (D) [3] |
|
| Cybrids (45–100%) |
(D) [3][35][37][40][44][45][46][47][48][49] | (D) [3]; (N) [40] |
(D) [29][40][46][47][48][50][51] | (A) [37][44][48] | Mitochondrial membrane potential (D) [50] or (I) [47][49]; Mitochondrial morphology (A) [52][53]; Mitochondrial ultrastructure (A) [49]; ROS (I) [47][50][54]; Antioxidant enzymes (A) [47][54]; Growth in galactose media (D) [35][45][47]; ATP level (D) [50]; Extracellular lactate (I) [3][46]; Autophagy (I) [53]; CI, CII, or CIV activities (D) [3][47][48]; Oligomycin [37] and apoptosis [49] sensitivity (I); Actin cytoskeleton and Ca2+ in-flux rates (A) [52]; Reductive carboxylation of glutamine and NADH/NAD ratio (I) [51][55] |
|
| IPSCs (90–100%) |
(D) [56]; (N) [57] | Mitochondrial membrane potential, ROS, and lactate production (I) [58] | ||||
| NPCs, Neurons (90–100%) |
(D) [58] | Mitochondrial membrane potential, ROS, and antioxidant enzymes (I) [58]; Degenerative defect [58]; Metabolic dysregulation; Formation of cerebral organoid (A) [57] |
||||
| m.8993T>C ATP6: p.L156P |
Yeast (100%) [59] |
(D) | (N) | (D) | (N) | CIV level, COX2, and ATP6 protein levels (D) |
| Lymphocytes (90–95%) |
(D) [31] | Mitochondrial membrane potential (N); ROS (I) [31]; Proton flux (D) [32] |
||||
| Fibroblasts (95–100%) |
(D) [3][34] | (D) [3], (N) [34] | (N) [34] | (N) [37] | Depolarized plasma membrane and ROS (I) [4]; Growth in galactose media (D) [3] |
|
| Cybrids (100%) |
(D) [40][46]; (N) [3][48] |
(N) [3] | (D) [46]; (N) [48] |
(N) [37][48] | Lactate production (I) [3][46] | |
| m.9008C>G ATP6: p.T161S |
Muscle (100%) [3] |
(N) | ||||
| Fibroblasts (100%) [3] |
(N) | (N) | CI activity (D) | |||
| Cybrids (100%) [3] |
(D) | (N) | Growth in galactose media (D); Lactate production (I) |
|||
| m.9016A>G ATP6: p.I164V |
Yeast (100%) [16] |
(N) | (N) | (N) | Mitochondrial membrane potential (N) | |
| m.9019A>G ATP6: p.T165A |
Muscle (100%) [3] |
(D) | CI activity (D) | |||
| m.9025G>A ATP6: p.G167S |
Yeast (100%) [16] |
(D) | (D) | (N) | Sensitivity of growth to oligomycin (I); Mitochondrial membrane potential (N) |
|
| m.9029A>G ATP6: p.H168R |
Yeast (100%) [16] |
(D) | (D) | (N) | Sensitivity of growth to oligomycin (I); Mitochondrial membrane potential (N) |
|
| Cybrids (100%) [50] |
(D) | ATP level (D); ROS and mitochondrial membrane potential (I) |
||||
| m.9032T>C ATP6: p.L169P |
Cybrids (25–80%) [50] |
(D) | ATP level (D); ROS and mitochondrial membrane potential (I) |
|||
| m.9035T>C ATP6: p.L170P |
Cybrids (100%) [60] |
(D) | ROS and antioxidant enzymes (I); Mitochondrial membrane potential (N); Sensitivity to glucose deprivation (I); Oxidative stress (I) |
|||
| Muscle (100%) [3] |
(D) | CI activity (D) | ||||
| Fibroblasts (100%) |
(D) [3] | (N) [3] | (D) [61] | (A) [61] | Growth in galactose media (D) [3] | |
| m.9058A>G ATP6: p.T178A |
Yeast (100%) [16] |
(N) | (N) | (N) | Mitochondrial membrane potential (N) | |
| m.9101T>C ATP6: p.I192T |
Lymphocytes (100%) [62][63] |
(D) | ||||
| Cybrids (100%) [63] |
(D) | |||||
| m.9127 delAT ATP6: p.I201PfsX2 |
Fibroblasts (50%) [64] |
(D) | (D) | (N) | Oligomycin-induced increase in mitochondrial membrane potential (D) | |
| m.9134A>G ATP6: p.E203G |
Muscle [65] | (D) | (D) | |||
| m.9139G>A ATP6: p.A205T |
Yeast (100%) [16] |
(N) | (N) | (N) | Mitochondrial membrane potential (N) | |
| m.9154C>T ATP6: p.Q210X |
Fibroblasts [66] | (N) | (A) | Mitochondrial morphology (A) | ||
| IPSC and Neurons [66] | (A) | Motor neuron differentiation (A); Mitochondrial morphology (A); Hyperactivation of the Notch pathway |
||||
| m.9160T>C ATP6: p.Y212H |
Yeast (100%) [16] |
(N) | (N) | (N) | Mitochondrial membrane potential (N) | |
| m.9176T>G ATP6: p.L217R |
Yeast (100%) [67] |
(D) | (D) | (A) | Growth in stress conditions (D); CIV super-complexes (D); ATP6, COX2, and CYTB protein levels (D); Mitochondrial ultrastrucure (A) |
|
| Muscle (>95%) [33] |
(A) | |||||
| Fibroblasts (95–100%) |
(D) [68] | (N) [40] | (N) [68] | Mitochondrial membrane potential (I) [68]; Growth in galactose media (D) [40] |
||
| Cybrids (30–100%) |
(D) [40][48][49] | (N) [40] | (D) [40][48] | (A) [48] | CI and CIV activities (D) [48]; Mitochondrial ultrastructure (A) [49]; Mitochondrial membrane potential and apoptosis sensitivity (I) [49] |
|
| m.9176T>C ATP6: p.L217P |
Yeast [69] | (D) | (N) | (D) | (A) | |
| Muscle (100%) [3] |
(D) | |||||
| Fibroblasts (100%) |
(N) [70]; (D) [71] | (N) [40] | (A) [71] | Mitochondrial network morphology (N) [71]; Depolarized plasma membrane and ROS (I) [4] |
||
| Cybrids (100%) [40] |
(D) | (N) | ||||
| m.9185T>C ATP6: p.L220P |
Yeast [72] | (D) | (N) | (N) | (N) | Sensitivity of growth to oligomycin (I) |
| Muscle (>97%) |
(D) [73][74] | (A) [74] | CI, CII, and CIV activities (N) [75][76][77] | |||
| Lymphocytes [75] |
(N) | (D) | ||||
| Fibroblasts (90–100%) |
(D) [3][4] | (D) [4]; (N) [3] | (D) [4] | (N) [4] | CI activity (D) and depolarized plasma membrane [4]; ROS or antioxidant enzymes (I) [4][78]; CI, CII, and CIV activities (N) [75]; Mitochondrial membrane potential (N) [78]; Lactate production (I) [3] |
|
| Cybrids (100%) | (D) [78]; (N) [3] |
(D) [4]; (N) [3] | (D) [4] | CI activity (D) [4]; Lactate production (I) [3]; Mitochondrial membrane potential (N) [78] |
||
| NPC and neuron (100%) [78] | (D) | (N) | Mitochondrial membrane potential (I); Mitochondrial calcium homeostasis (A); Depolarized plasma membrane; Mitochondrial cristae structure and ROS (N) |
|||
| m.9191T>C ATP6: p.L222P |
Muscle (94%) [73] |
(D) | (D) | |||
| Yeast [72][79] |
(D) | (N) | (D) | (A) | Growth in stress conditions (D); CIV level (D); ATP6 protein level (D) |
|
| m.9205delTA ATP6: p.X227NA |
Muscle (>98%) [80] |
CIV activity (D) | ||||
| Fibroblasts (>98%) |
(D) [80] | (N) [80] | (D) [80] | (A) [80] | CIV activity (D) [80][81]; ATP6 protein and CIV subunit levels (D) [80]; Morphological abnormalities of mitochondria [81] |
|
2. The mt-DNA Pathogenic Variants at Position m.8993
The two most common variants in MT-ATP6 are m.8993T>G (p.Leu156Arg) and m.8993T>C (p.Leu156Pro), which cause a change in a highly conserved leucine residue on ATP6 [26][82][83]. These variants are the most common and are responsible for approximately 50% of reported MT-ATP6 disease cases [84]. These variants are associated with Neuropathy, Ataxia, and Retinitis Pigmentosa (NARP) or Maternal Inherited Leigh syndrome (MILS) when heteroplasmy is between 70 and 90% or greater than 90%, respectively. Furthermore, the T>G transversion usually results in a more severe clinical phenotype than the T>C transition [82][83][85][86].
2.1. Biochemical and Cellular Dysfunctions in Mutated Cell Models
Analyses of patient specimens carrying the m.8993T>G or the m.8993T>C variant have been performed in platelets [26][27], lymphocytes [28][29][30][31][32], muscle tissue [33], and skin fibroblasts [3][4][34][35][36][37][38][39][40][41][42][43][87] (see Table 1). Several biochemical abnormalities have been identified, including a decreased ATP synthesis and oxygen consumption rate (OCR) [3][26][27][28][29][30][31][34][35][36][37][38][39][40][41][42], often in direct correlation with the mutation load [27][30][40][45], an alteration of the proton flux [30][31][32], and a not-fully assembled CV [33]. In human cells, these ATP synthase dysfunctions lead, as secondary effects, to a reduction in growth in stress medium [3][35][38][40], to an increase in both mitochondrial membrane potential (MMP) [31][36][39][87] and ROS [4][31][39][43], as well as to an altered mitochondrial network morphology and cristae structure [39][42].
In cells of NARP patients carrying the m.8993T>G variant, the severe impairment of OXPHOS has been proposed as the primary pathogenic defect; instead, the increase in ROS could be the main contributor to the pathogenesis of the disease associated with the m.8993T>C variant [31].
It is worth noting that no significant effects of the m.8993T>G variant have been reported on either ATP hydrolytic activity or ATP-driven proton transport by Complex V in patient cells [26][36]. However, inhibition of ATP hydrolytic activity could contribute to energy preservation and survival of cells under stress conditions (oxygen shortage), leading to the collapse of the proton motive force and ATP synthase working in reverse. Incidentally, due to the heterogeneity of the membrane potential within the same mitochondrion and the possible coexistence of ATP synthase working physiologically and in reverse [88][89], patients might benefit from the use of a specific inhibitor of the hydrolytic activity of CV.
The different percentages of heteroplasmy and other factors, including nuclear background and the type of tissue analyzed, may contribute to the phenotypic differences observed in the analysis of patients’ specimens, as well as those observed in clinical outcomes [61][84][90]. For these reasons, transmitochondrial cybrids are widely used to validate the possible pathogenicity of a mitochondrial variant, even in homoplasmic populations, with the advantage of using a cell model with the same nuclear background [91]. In the case of the two MT-ATP6 variants, this cell model clearly demonstrated the impairment of respiration [29][40][46][47][48][50][51], ATP synthesis [3][35][37][40][44][45][46][47][48][49], mitochondrial morphology [52][53], and enhanced ROS production [47][50][54], confirming the milder effect of m.8993T>C compared to the T>G variant [31][37][40][46][48].
Interestingly, analysis of different cybrid lines carrying the same m.8993T>G variant highlighted that the mitochondrial genome sequence, and thus the haplogroup, is a factor contributing to the variations in the observed biochemical phenotypes, ranging from normal to severe defects [48]. Moreover, clear evidence of the role of mutation load on deleterious biochemical abnormalities has been recently reported in isogenic cybrids, where the OCR reduction and the extracellular acidification rate (ECAR) increase were proportionally linked to the levels of heteroplasmy, indicating a switch toward glycolysis [51]. Metabolic remodeling induced by the m.8993T>G variant was also investigated, and both proteomics and metabolomics analysis were consistent with increased glycolysis and reductive carboxylation of glutamine to support cell survival and to maintain redox balance [51]. Accordingly, a second report showed that, in cybrids, the impaired OXPHOS activity induces compensatory energy-generating anaplerotic mechanisms where glutamine-glutamate-α-ketoglutarate metabolism sustains cell survival [55].
The deleterious mechanism hypothesized based on all these studies, especially for the m.8993T>G variant, includes defective proton transport across Fo, failure of the enzyme to couple phosphorylation of ADP on F1 to proton flow, or alteration of the holoenzyme assembly and stability [30][31][32][84]. Considering that these alterations have been observed in different cellular models despite not always being together, it seems reasonable that all three mechanisms contribute to the pathogenicity of the variants.
In recent years, the introduction of induced pluripotent stem cell (iPSC) technology allowed disease modeling by overcoming the difficulty of accessing clinically relevant patients’ cells or tissues, such as neurons. The generation of IPSCs requires multiple quality checks and presents the issue of heteroplasmy fluctuations due to the genetic bottleneck occurring in the reprogramming process. In addition, the mutant load can also change during differentiation or cell culture and therefore must be constantly monitored [92][93].
A series of patient-derived iPSCs carrying the m.8993T>G or T>C variant has been developed [58][94][95][96][97], and neural progenitor cells (NPCs) and neurons have been differentiated [58]. The mutant iPSCs were able to differentiate into the three embryonic germ layers (endoderm, mesoderm, and ectoderm) [78][96][97]. However, analysis of embryoid bodies showed impaired differentiation potential in cells with a high percentage of the variant [96]. Overall, the generated cell types recapitulate the energy defects observed in other cell models and the degenerative phenotypes observed in patients [56][58]. Neurons, in part because of their predominantly mitochondria-dependent oxidative metabolism [58][78], have shown degenerative defects not detectable in other, less differentiated cells, notably ATP shortage and AMPK activation, finer neuronal fibers, and increased sensitivity to glutamate toxicity [58]. Furthermore, a study of mutant IPSCs revealed abnormalities during the three-dimensional differentiation and a defective formation of cerebral organoids, particularly in the generation of neural epithelial buds, as well as impaired corticogenesis with an altered metabolic profile [57].
2.2. Modeling of ATP6 Subunit Carrying Changes in Leu156 in the ATP Synthase Human Structure
In the recently released ATP synthase human structures ([98], PDB id 8H9S, 8H9T, 8H9U, and 8H9V for states 1, 2, 3a, and 3b, respectively), the Leu156 residue is located on helix H5 and is buried in the core of ATP6, forming van der Waals contacts with Leu217 and Val218, located on helix H6 (Figure 1). In the four structures, the residues in the vicinity of Leu156 are the same, and there are no conformational transitions involving ATP6 in the different states of the human ATP synthase. The variant of Leu156 in arginine can damage the interaction between subunit helices H5 and H6 because of (i) the larger volume of an arginine residue with respect to a leucine and (ii) the presence of a charged side chain in a region populated only by hydrophobic residues. In other words, the presence of an arginine in position 156 can cause a divarication between helices H5 and H6 that in turn can alter the folding of ATP6. Indeed, the correct positioning of these helices is crucial for proton access to the negatively charged Glu58 in the c subunits (Figure 1).
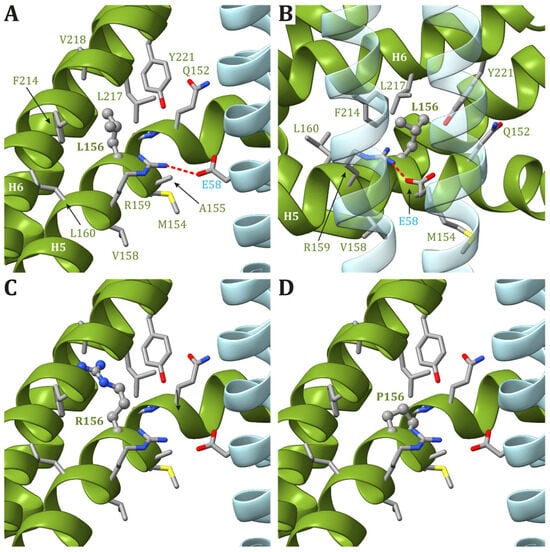
Figure 1. Detail of the region comprising Leu156 of ATP6 in the human structure of ATP synthase (state 1). (A,B) The native ATP6 and c subunits are reported in ribbons colored as in Figure 2. Residues labels are colored as the corresponding subunits. Leu156 is in ball-and-stick, while other residues in the vicinity of Leu156 or proposed to be part of the proton translocation process are in stick. The side chains are colored according to the atom type. The interaction between Arg159 in ATP6 and Glu58 in c8-ring is shown. The orientation of panel (B) is clockwise rotated by 90° around the vertical axis with respect to the orientation in panel (A). Panels (C,D) reports the model structures of the p.Leu156Arg and p.Leu156Pro variants, respectively.
3. The Pathogenic Variants at Nucleotide m.9176
The m.9176T>C and m.9176T>G variants, which cause a Leu217Pro and a Leu217Arg ammino acid change, are frequently found in MILS patients and were first reported in 1995 and 2001, respectively [68][70].
In the case of the m.9176T>G variant, a partially disassembled CV, decreased ATP synthesis, and mitochondrial respiration due to a defective OXPHOS pathway led to an increase in MMP in MILS patient-derived fibroblasts [40][68]. Similar observations have been described in cybrids [40][48][49] and in patient-derived muscle tissue [33]. Furthermore, analysis of this variant in yeast highlighted a severe reduction in the ATP6 protein level, suggesting that it may affect the assembly of the ATP synthase complex and cause mitochondrial and bioenergetic dysfunctions [67]. Human IPSCs have been recently generated for the m.9176T>G [97], and their use to develop neurons will be instrumental in deeply characterizing the pathogenic mechanism of this variant in a disease-target tissue.
In the first report in 1995, biochemical analysis of the m.9176T>C variant revealed no defects in ATP synthase function in patient cells with the homoplasmic variant [70]. However, further studies reported impaired ATP synthesis [40][69][71] and CV stability [71] in both human cells and mutant yeast. As for the variants at the nucleotide m.8993, the alteration caused by the T>C variant was less severe than that caused by the T>G variant [40][69].
In human structures, Leu217 is positioned in helix H6 of ATP6 and is part of both the interface between helices H5 and H6 and between ATP6 and the c8-ring. While the residues in ATP6 in the vicinity of Leu217 (Leu156, Arg159, Val213, Leu216, Leu220, and Tyr221) do not change during the catalytic cycle, the residues found close to Leu217 in the c subunit are different depending on the ATP synthase state. Indeed, in state 1, Leu52 and Leu56 in the c subunit are close to Leu217 in ATP6 (Figure 2A), while in states 2 and 3a, Leu217 is in the vicinity of Leu52 and Ala55 in the c subunit (Figure 2B). Finally, in state 3b, no residue from any c subunit is in the closeness of Leu217 (Figure 2C). The variant of Leu217 in an arginine residue appears to cause two effects: (i) arginine is a larger residue with respect to leucine, causing some sort of friction between the ATP6 and the c8-ring that is rotating during the catalytic cycle, and (ii) the arginine has a charged side chain that can form H-bonds with other residues in the vicinity, such as Tyr221 from ATP6. This newly formed H-bond can interfere with the formation of another H-bond between Tyr221 and a water molecule in the outlet proton translocation half-channel. The latter water molecule is held in the correct position by two H-bonds, the already cited one with Tyr221 and a second with Glu58 from the c subunit. On the other hand, the variant of Leu217 in a proline residue can cause some sort of effects on the folding of helix H6 downstream of the mutated residue, but—as in the case of p.Leu156Pro—the damage caused by the presence of a small hydrophobic residue in position 156 should be moderate.
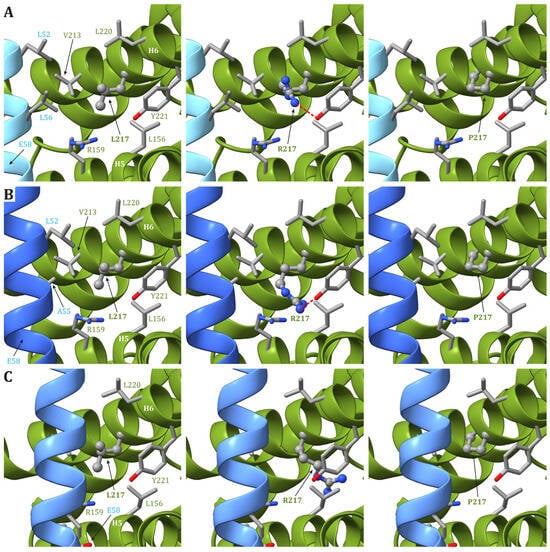
Figure 2. Detail of the region comprising Leu217 from ATP6 in the human structure of ATP synthase in state 1 (A), state 2 and 3a (B), and 3b (C). The native ATP6 and c subunits are reported in ribbons colored as in Figure 2. Residue labels are colored as the corresponding subunits. Leu217 is shown as ball-and-stick, while other residues in the vicinity of Leu217 or proposed to be part of the proton translocation process are shown as a stick. The side chains are colored according to the atom type. In the left panels, the wild-type protein is reported, while the models of Leu217Arg and Leu217Pro variants are reported in the central and right panels, respectively. H-bonds are indicated using dashed red lines.
4. The mt-DNA Pathogenic Variant at Position m.9185
The variant at nucleotide m.9185T>C (p.Leu220Pro) was first reported in 2005 [73], and functional studies revealed a moderate effect of this variant on ATP synthase functioning. Indeed, in patient cells, besides normal Complexes I-IV activity [73][76][77], a slight alteration of CV and a depolarization of the plasma membrane were reported, often only in the case of homoplasmy [3][4][73][74][75][78]. Mild effects on ATPase function have also been observed in mutated cybrids and yeast cells [3][4][72][78]. The evaluation of IPSCs and their patient-derived NPCs [78][99], in addition to the defective ATP production, allowed people to highlight mitochondrial impairment that is hidden in the other cell types. Indeed, neural cells presented a mitochondrial hyperpolarization and an alteration of mitochondrial calcium homeostasis, as evidenced by both transcriptomic and proteomic analysis [78]. All these data suggest that the variant may alter the ability of these cells to produce ATP and control MMP, causing neural impairment.
In the human ATP synthase structures, Leu220 is located on helix H6 of ATP6, just one helix turn away from Leu217, and forms van der Waals contacts with Met60, located on helix H3. Except for Glu224, the other residues found in the vicinity of Leu220 in ATP6 are all hydrophobic (Leu216, Leu217, and Tyr221). As for Leu217, Leu220 is at the interface between ATP6 and the c8-ring, and the interacting residues from the latter depend on the ATP synthase state. Leu220 of ATP6 is in the vicinity of some residues of the c subunit: Phe47 and Ile51 in states 1 and 3b (Figure 3A) and Leu52 in states 2 and 3a (Figure 3B). As for the previously discussed cases, a variant of Leu220 in a proline residue can have some effects on the fold of helix H6 downstream of the mutated residue and, in turn, cause some problems to the ATP synthase mechanism. On the other hand, proline is a small hydrophobic residue, and no serious steric or electrostatic effects are expected.
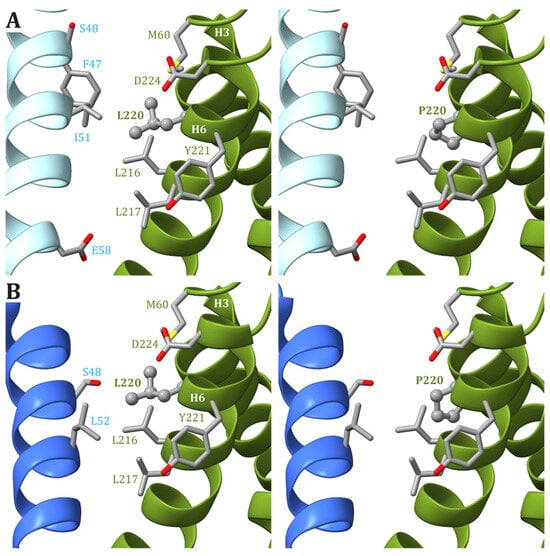
Figure 3. Detail of the region comprising Leu220 from ATP6 in the human structure of ATP synthase in states 1 and 3b (A) and states 2 and 3a (B). The native ATP6 and c subunits are reported in ribbons colored as in Figure 2. Residue labels are colored as the corresponding subunits. Leu220 is shown as ball-and-stick, while other residues in the vicinity of Leu220 or proposed to be part of the proton translocation process are shown as a stick. The side chains are colored according to the atom type. In the left panels, the wild-type protein is reported, while the models of Leu220Pro variants are in the right panels.
5. Other MT-ATP6 and MT-ATP8 Pathogenic Variants
A number of MT-ATP6 and MT-ATP8 variants reported in the literature have been reviewed by two different research groups [82][84], and the functional studies of cell models are reported in detail in Table 1.
The study of the m.8909T>C variant, found in a patient also carrying the pathogenic m.3243A>G variant in mt-tRNALeu (MT-TL1), reported a defect in Complex V assembly and ATP synthesis [18]. A compromised assembly of ATP synthase and a reduced OCR has been observed in fibroblasts of two patients carrying the same m.8782G>A variant, one presenting adult-onset cerebellar ataxia, chronic kidney disease, and diabetes, whereas the other had myoclonic epilepsy and cerebellar ataxia [14].
The truncating variant m.9154C>T was found in a patient with adult-onset axonal neuropathy, ataxia, and IgA nephropathy and caused alteration of Complex V assembly, mitochondrial morphology, and ultrastructure in mutated fibroblasts [66]. Interestingly, the mutation load resulted to be proportional to Complex V assembly defect in patient-derived iPSCs and responsible for impaired neurogenesis due to Notch hyperactivation and altered metabolism of mature motor neurons [66].
Other identified MT-ATP6 variants include the m.8858G>A variant in a sporadic case of NARP-MILS [100]; the m.8936T>A in a young boy with atypical mitochondrial Leigh syndrome associated with bilateral basal ganglia calcifications [101]; m.9143T>C in a patient with insulin-dependent diabetes mellitus, recurrent lactic acidosis, infections, and immunodeficiency [102]; m.9154C>T in a patient with neuropathy, cerebellar ataxia, and IgA nephropathy [103]; and m.9171A>G in a patient with mitochondrial retinopathy with atrophy [104]. The three variants m.8572G>A, the m.8578C>T and m.8812A>G were found in patients with adult-onset spinocerebellar ataxia (SCA) [105].
Regarding new variants affecting MT-ATP6, MT-ATP8, or both, m.8561C>T, which causes a defect in CV assembly, was reported in a child with early onset ataxia, psychomotor delay, and microcephaly [11], whereas functional studies have been performed for the m.8382C>T, m.8424T>C, m.8806C >G, m.8975T>C, m.9008C>G, and m.9019A>G variants [3].
References
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160.
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Leeuwenburgh, C.; Bucci, C.; Marzetti, E. The Contribution of Mitochondrial DNA Alterations to Aging, Cancer, and Neurodegeneration. Exp. Gerontol. 2023, 178, 112203.
- Rucheton, B.; Jardel, C.; Filaut, S.; Amador, M.D.M.; Maisonobe, T.; Serre, I.; Romero, N.B.; Leonard-Louis, S.; Haraux, F.; Lombès, A. Homoplasmic Deleterious MT-ATP6/8 Mutations in Adult Patients. Mitochondrion 2020, 55, 64–77.
- Aure, K.; Dubourg, O.; Jardel, C.; Clarysse, L.; Sternberg, D.; Fournier, E.; Laforet, P.; Streichenberger, N.; Petiot, P.; Gervais-Bernard, H.; et al. Episodic Weakness Due to Mitochondrial DNA MT-ATP6/8 Mutations. Neurology 2013, 81, 1810–1818.
- Panja, C.; Niedzwiecka, K.; Baranowska, E.; Poznanski, J.; Kucharczyk, R. Analysis of MT-ATP8 Gene Variants Reported in Patients by Modeling in Silico and in Yeast Model Organism. Sci. Rep. 2023, 13, 9972.
- Ware, S.M.; El-Hassan, N.; Kahler, S.G.; Zhang, Q.; Ma, Y.W.; Miller, E.; Wong, B.; Spicer, R.L.; Craigen, W.J.; Kozel, B.A.; et al. Infantile Cardiomyopathy Caused by a Mutation in the Overlapping Region of Mitochondrial ATPase 6 and 8 Genes. J. Med. Genet. 2009, 46, 308–314.
- Imai, A.; Fujita, S.; Kishita, Y.; Kohda, M.; Tokuzawa, Y.; Hirata, T.; Mizuno, Y.; Harashima, H.; Nakaya, A.; Sakata, Y.; et al. Rapidly Progressive Infantile Cardiomyopathy with Mitochondrial Respiratory Chain Complex V Deficiency Due to Loss of ATPase 6 and 8 Protein. Int. J. Cardiol. 2016, 207, 203–205.
- Jonckheere, A.I.; Hogeveen, M.; Nijtmans, L.G.J.; Van Den Brand, M.A.M.; Janssen, A.J.M.; Diepstra, J.H.S.; Van Den Brandt, F.C.A.; Van Den Heuvel, L.P.; Hol, F.A.; Hofste, T.G.J.; et al. A Novel Mitochondrial ATP8 Gene Mutation in a Patient with Apical Hypertrophic Cardiomyopathy and Neuropathy. J. Med. Genet. 2007, 45, 129–133.
- Boominathan, A.; Vanhoozer, S.; Basisty, N.; Powers, K.; Crampton, A.L.; Wang, X.; Friedricks, N.; Schilling, B.; Brand, M.D.; O’Connor, M.S. Stable Nuclear Expression of ATP8 and ATP6 Genes Rescues a mtDNA Complex V Null Mutant. Nucleic Acids Res. 2016, 44, 9342–9357.
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A Novel Mutation m.8561C>G in MT-ATP6/8 Causing a Mitochondrial Syndrome with Ataxia, Peripheral Neuropathy, Diabetes Mellitus, and Hypergonadotropic Hypogonadism. J. Neurol. 2016, 263, 2188–2195.
- Fragaki, K.; Chaussenot, A.; Serre, V.; Acquaviva, C.; Bannwarth, S.; Rouzier, C.; Chabrol, B.; Paquis-Flucklinger, V. A Novel Variant m.8561C>T in the Overlapping Region of MT-ATP6 and MT-ATP8 in a Child with Early-Onset Severe Neurological Signs. Mol. Genet. Metab. Rep. 2019, 21, 100543.
- Jackson, C.B.; Hahn, D.; Schröter, B.; Richter, U.; Battersby, B.J.; Schmitt-Mechelke, T.; Marttinen, P.; Nuoffer, J.-M.; Schaller, A. A Novel Mitochondrial ATP6 Frameshift Mutation Causing Isolated Complex V Deficiency, Ataxia and Encephalomyopathy. Eur. J. Med. Genet. 2017, 60, 345–351.
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; Lopez-Perez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP Syndrome in a Patient Harbouring an Insertion in the MT-ATP6 Gene That Results in a Truncated Protein. J. Med. Genet. 2008, 46, 64–67.
- Bugiardini, E.; Bottani, E.; Marchet, S.; Poole, O.V.; Beninca, C.; Horga, A.; Woodward, C.; Lam, A.; Hargreaves, I.; Chalasani, A.; et al. Expanding the Molecular and Phenotypic Spectrum of Truncating MT-ATP6 Mutations. Neurol. Genet. 2020, 6, e381.
- Blanco-Grau, A.; Bonaventura-Ibars, I.; Coll-Cantí, J.; Melià, M.J.; Martinez, R.; Martínez-Gallo, M.; Andreu, A.L.; Pinós, T.; García-Arumí, E. Identification and Biochemical Characterization of the Novel Mutation m. 8839G>C in the Mitochondrial ATP6 Gene Associated with NARP Syndrome. Genes Brain Behav. 2013, 12, 812–820.
- Baranowska, E.; Niedzwiecka, K.; Panja, C.; Charles, C.; Dautant, A.; Poznanski, J.; Di Rago, J.-P.; Tribouillard-Tanvier, D.; Kucharczyk, R. Probing the Pathogenicity of Patient-Derived Variants of MT-ATP6 in Yeast. Dis. Models Mech. 2023, 16, dmm049783.
- Kucharczyk, R.; Giraud, M.-F.; Brèthes, D.; Wysocka-Kapcinska, M.; Ezkurdia, N.; Salin, B.; Velours, J.; Camougrand, N.; Haraux, F.; Di Rago, J.-P. Defining the Pathogenesis of Human mtDNA Mutations Using a Yeast Model: The Case of T8851C. Int. J. Biochem. Cell Biol. 2013, 45, 130–140.
- Ding, Q.; Kucharczyk, R.; Zhao, W.; Dautant, A.; Xu, S.; Niedzwiecka, K.; Su, X.; Giraud, M.-F.; Gombeau, K.; Zhang, M.; et al. Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase. Life 2020, 10, 215.
- Niedzwiecka, K.; Kabala, A.M.; Lasserre, J.-P.; Tribouillard-Tanvier, D.; Golik, P.; Dautant, A.; Di Rago, J.-P.; Kucharczyk, R. Yeast Models of Mutations in the Mitochondrial ATP6 Gene Found in Human Cancer Cells. Mitochondrion 2016, 29, 7–17.
- Abu-Amero, K.K.; Bosley, T.M. Detection of Mitochondrial Respiratory Dysfunction in Circulating Lymphocytes Using Resazurin. Arch. Pathol. Lab. Med. 2005, 129, 1295–1298.
- Wen, S.; Niedzwiecka, K.; Zhao, W.; Xu, S.; Liang, S.; Zhu, X.; Xie, H.; Tribouillard-Tanvier, D.; Giraud, M.-F.; Zeng, C.; et al. Identification of G8969>A in Mitochondrial ATP6 Gene That Severely Compromises ATP Synthase Function in a Patient with IgA Nephropathy. Sci. Rep. 2016, 6, 36313.
- Skoczeń, N.; Dautant, A.; Binko, K.; Godard, F.; Bouhier, M.; Su, X.; Lasserre, J.-P.; Giraud, M.-F.; Tribouillard-Tanvier, D.; Chen, H.; et al. Molecular Basis of Diseases Caused by the mtDNA Mutation m.8969G>A in the Subunit a of ATP Synthase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 602–611.
- Burrage, L.C.; Tang, S.; Wang, J.; Donti, T.R.; Walkiewicz, M.; Luchak, J.M.; Chen, L.-C.; Schmitt, E.S.; Niu, Z.; Erana, R.; et al. Mitochondrial Myopathy, Lactic Acidosis, and Sideroblastic Anemia (MLASA) plus Associated with a Novel de Novo Mutation (m.8969G>A) in the Mitochondrial Encoded ATP6 Gene. Mol. Genet. Metab. 2014, 113, 207–212.
- Duno, M.; Wibrand, F.; Baggesen, K.; Rosenberg, T.; Kjaer, N.; Frederiksen, A.L. A Novel Mitochondrial Mutation m.8989G>C Associated with Neuropathy, Ataxia, Retinitis Pigmentosa—The NARP Syndrome. Gene 2013, 515, 372–375.
- Rak, M.; Tetaud, E.; Duvezin-Caubet, S.; Ezkurdia, N.; Bietenhader, M.; Rytka, J.; Di Rago, J.-P. A Yeast Model of the Neurogenic Ataxia Retinitis Pigmentosa (NARP) T8993G Mutation in the Mitochondrial ATP Synthase-6 Gene. J. Biol. Chem. 2007, 282, 34039–34047.
- Baracca, A.; Barogi, S.; Carelli, V.; Lenaz, G.; Solaini, G. Catalytic Activities of Mitochondrial ATP Synthase in Patients with Mitochondrial DNA T8993G Mutation in the ATPase 6 Gene Encoding Subunit a. J. Biol. Chem. 2000, 275, 4177–4182.
- Carelli, V.; Baracca, A.; Barogi, S.; Pallotti, F.; Valentino, M.L.; Montagna, P.; Zeviani, M.; Pini, A.; Lenaz, G.; Baruzzi, A.; et al. Biochemical-Clinical Correlation in Patients with Different Loads of the Mitochondrial DNA T8993G Mutation. Arch. Neurol. 2002, 59, 264.
- Tatuch, Y.; Robinson, B.H. The Mitochondrial DNA Mutation at 8993 Associated with NARP Slows the Rate of ATP Synthesis in Isolated Lymphoblast Mitochondria. Biochem. Biophys. Res. Commun. 1993, 192, 124–128.
- Trounce, I.; Neill, S.; Wallace, D.C. Cytoplasmic Transfer of the mtDNA Nt 8993 T-->G (ATP6) Point Mutation Associated with Leigh Syndrome into mtDNA-Less Cells Demonstrates Cosegregation with a Decrease in State III Respiration and ADP/O Ratio. Proc. Natl. Acad. Sci. USA 1994, 91, 8334–8338.
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient Coupling between Proton Transport and ATP Synthesis May Be the Pathogenic Mechanism for NARP and Leigh Syndrome Resulting from the T8993G Mutation in mtDNA. Biochem. J. 2006, 395, 493–500.
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical Phenotypes Associated with the Mitochondrial ATP6 Gene Mutations at Nt8993. Biochim. Biophys. Acta (BBA)-Bioenerg. 2007, 1767, 913–919.
- Solaini, G.; Harris, D.A.; Lenaz, G.; Sgarbi, G.; Baracca, A. The Study of the Pathogenic Mechanism of Mitochondrial Diseases Provides Information on Basic Bioenergetics. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1777, 941–945.
- Carrozzo, R.; Wittig, I.; Santorelli, F.M.; Bertini, E.; Hofmann, S.; Brandt, U.; Schägger, H. Subcomplexes of Human ATP Synthase Mark Mitochondrial Biosynthesis Disorders. Ann. Neurol. 2006, 59, 265–275.
- Vázquez-Memije, M.E.; Shanske, S.; Santorelli, F.M.; Kranz-Eble, P.; DeVivo, D.C.; DiMauro, S. Comparative Biochemical Studies of ATPases in Cells from Patients with the T8993G or T8993C Mitochondrial DNA Mutations. J. Inherit. Metab. Dis. 1998, 21, 829–836.
- Manfredi, G.; Gupta, N.; Vazquez-Memije, M.E.; Sadlock, J.E.; Spinazzola, A.; De Vivo, D.C.; Schon, E.A. Oligomycin Induces a Decrease in the Cellular Content of a Pathogenic Mutation in the Human Mitochondrial ATPase 6 Gene. J. Biol. Chem. 1999, 274, 9386–9391.
- García, J.J.; Ogilvie, I.; Robinson, B.H.; Capaldi, R.A. Structure, Functioning, and Assembly of the ATP Synthase in Cells from Patients with the T8993G Mitochondrial DNA Mutation. J. Biol. Chem. 2000, 275, 11075–11081.
- Cortés-Hernández, P.; Vázquez-Memije, M.E.; García, J.J. ATP6 Homoplasmic Mutations Inhibit and Destabilize the Human F1F0-ATP Synthase without Preventing Enzyme Assembly and Oligomerization. J. Biol. Chem. 2007, 282, 1051–1058.
- Bonnet, C.; Kaltimbacher, V.; Ellouze, S.; Augustin, S.; Bénit, P.; Forster, V.; Rustin, P.; Sahel, J.-A.; Corral-Debrinski, M. Allotopic mRNA Localization to the Mitochondrial Surface Rescues Respiratory Chain Defects in Fibroblasts Harboring Mitochondrial DNA Mutations Affecting Complex I or V Subunits. Rejuvenation Res. 2007, 10, 127–144.
- Lebiedzinska, M.; Karkucinska-Wieckowska, A.; Wojtala, A.; Suski, J.M.; Szabadkai, G.; Wilczynski, G.; Wlodarczyk, J.; Diogo, C.V.; Oliveira, P.J.; Tauber, J.; et al. Disrupted ATP Synthase Activity and Mitochondrial Hyperpolarisation-Dependent Oxidative Stress Is Associated with p66Shc Phosphorylation in Fibroblasts of NARP Patients. Int. J. Biochem. Cell Biol. 2013, 45, 141–150.
- Vazquez-Memije, M.E.; Rizza, T.; Meschini, M.C.; Nesti, C.; Santorelli, F.M.; Carrozzo, R. Cellular and Functional Analysis of Four Mutations Located in the Mitochondrial ATPase6 Gene. J Cell. Biochem. 2009, 106, 878–886.
- Mizuguchi, Y.; Hatakeyama, H.; Sueoka, K.; Tanaka, M.; Goto, Y. Low Dose Resveratrol Ameliorates Mitochondrial Respiratory Dysfunction and Enhances Cellular Reprogramming. Mitochondrion 2017, 34, 43–48.
- Uittenbogaard, M.; Brantner, C.A.; Fang, Z.; Wong, L.-J.C.; Gropman, A.; Chiaramello, A. Novel Insights into the Functional Metabolic Impact of an Apparent de Novo m.8993T>G Variant in the MT-ATP6 Gene Associated with Maternally Inherited Form of Leigh Syndrome. Mol. Genet. Metab. 2018, 124, 71–81.
- Geromel, V. Superoxide-Induced Massive Apoptosis in Cultured Skin Fibroblasts Harboring the Neurogenic Ataxia Retinitis Pigmentosa (NARP) Mutation in the ATPase-6 Gene of the Mitochondrial DNA. Hum. Mol. Genet. 2001, 10, 1221–1228.
- Nijtmans, L.G.J.; Henderson, N.S.; Attardi, G.; Holt, I.J. Impaired ATP Synthase Assembly Associated with a Mutation in the Human ATP Synthase Subunit 6 Gene. J. Biol. Chem. 2001, 276, 6755–6762.
- Manfredi, G.; Fu, J.; Ojaimi, J.; Sadlock, J.E.; Kwong, J.Q.; Guy, J.; Schon, E.A. Rescue of a Deficiency in ATP Synthesis by Transfer of MTATP6, a Mitochondrial DNA-Encoded Gene, to the Nucleus. Nat. Genet. 2002, 30, 394–399.
- Pallotti, F.; Baracca, A.; Hernandez-Rosa, E.; Walker, W.F.; Solaini, G.; Lenaz, G.; Melzi d’Eril, G.V.; Dimauro, S.; Schon, E.A.; Davidson, M.M. Biochemical Analysis of Respiratory Function in Cybrid Cell Lines Harbouring Mitochondrial DNA Mutations. Biochem. J. 2004, 384, 287–293.
- Mattiazzi, M. The mtDNA T8993G (NARP) Mutation Results in an Impairment of Oxidative Phosphorylation That Can Be Improved by Antioxidants. Hum. Mol. Genet. 2004, 13, 869–879.
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA Background Modifies the Bioenergetics of NARP/MILS ATP6 Mutant Cells. Hum. Mol. Genet. 2010, 19, 374–386.
- Carrozzo, R.; Rizza, T.; Lucioli, S.; Pierini, R.; Bertini, E.; Santorelli, F. A Mitochondrial ATPase 6 Mutation Is Associated with Leigh Syndrome in a Family and Affects Proton Flow and Adenosine Triphosphate Output When Modeled in Escherichia coli. Acta Paediatr. 2004, 93, 65–67.
- López-Gallardo, E.; Emperador, S.; Solano, A.; Llobet, L.; Martín-Navarro, A.; López-Pérez, M.J.; Briones, P.; Pineda, M.; Artuch, R.; Barraquer, E.; et al. Expanding the Clinical Phenotypes of MT-ATP6 Mutations. Hum. Mol. Genet. 2014, 23, 6191–6200.
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593.e7.
- Szczepanowska, J.; Zabłocki, K.; Duszyński, J. Influence of a Mitochondrial Genetic Defect on Capacitative Calcium Entry and Mitochondrial Organization in the Osteosarcoma Cells. FEBS Lett. 2004, 578, 316–322.
- Walczak, J.; Partyka, M.; Duszyński, J.; Szczepanowska, J. Implications of Mitochondrial Network Organization in Mitochondrial Stress Signalling in NARP Cybrid and Rho0 Cells. Sci. Rep. 2017, 7, 14864.
- Wojewoda, M.; Duszyński, J.; Szczepanowska, J. Antioxidant Defence Systems and Generation of Reactive Oxygen Species in Osteosarcoma Cells with Defective Mitochondria: Effect of Selenium. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 890–896.
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V.; et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab. 2018, 27, 1007–1025.e5.
- Ma, H.; Folmes, C.D.L.; Wu, J.; Morey, R.; Mora-Castilla, S.; Ocampo, A.; Ma, L.; Poulton, J.; Wang, X.; Ahmed, R.; et al. Metabolic Rescue in Pluripotent Cells from Patients with mtDNA Disease. Nature 2015, 524, 234–238.
- Romero-Morales, A.I.; Robertson, G.L.; Rastogi, A.; Rasmussen, M.L.; Temuri, H.; McElroy, G.S.; Chakrabarty, R.P.; Hsu, L.; Almonacid, P.M.; Millis, B.A.; et al. Human iPSC-Derived Cerebral Organoids Model Features of Leigh Syndrome and Reveal Abnormal Corticogenesis. Development 2022, 149, dev199914.
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of Neuronal Energy Deficiency by mTOR Inhibition as a Treatment for Mitochondria-Related Neurodegeneration. eLife 2016, 5, e13378.
- Kucharczyk, R.; Rak, M.; Di Rago, J.-P. Biochemical Consequences in Yeast of the Human Mitochondrial DNA 8993T>C Mutation in the ATPase6 Gene Found in NARP/MILS Patients. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 817–824.
- Sikorska, M.; Sandhu, J.K.; Simon, D.K.; Pathiraja, V.; Sodja, C.; Li, Y.; Ribecco-Lutkiewicz, M.; Lanthier, P.; Borowy-Borowski, H.; Upton, A.; et al. Identification of Ataxia-associated mtDNA Mutations (m.4052T>C and m.9035T>C) and Evaluation of Their Pathogenicity in Transmitochondrial Cybrids. Muscle Nerve 2009, 40, 381–394.
- Capiau, S.; Smet, J.; De Paepe, B.; Yildiz, Y.; Arslan, M.; Stevens, O.; Verschoore, M.; Stepman, H.; Seneca, S.; Vanlander, A. Clinical Heterogeneity in MT-ATP6 Pathogenic Variants: Same Genotype—Different Onset. Cells 2022, 11, 489.
- Lamminen, T.; Majander, A.; Juvonen, V.; Wikström, M.; Aula, P.; Nikoskelainen, E.; Savontous, M.L. A Mitochondrial Mutation at Nt 9101 in the ATP Synthase 6 Gene Associated with Deficient Oxidative Phosphorylation in a Family with Leber Hereditary Optic Neuroretinopathy. Am. J. Hum. Genet. 1995, 56, 1238–1240.
- Majander, A.; Lamminen, T.; Juvonen, V.; Aula, P.; Nikoskelainen, E.; Savontaus, M.-L.; Wikström, M. Mutations in Subunit 6 of the F1F0-ATP Synthase Cause Two Entirely Different Diseases. FEBS Lett. 1997, 412, 351–354.
- Mordel, P.; Schaeffer, S.; Dupas, Q.; Laville, M.-A.; Gérard, M.; Chapon, F.; Allouche, S. A 2 Bp Deletion in the Mitochondrial ATP 6 Gene Responsible for the NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa) Syndrome. Biochem. Biophys. Res. Commun. 2017, 494, 133–137.
- Honzik, T.; Tesarova, M.; Magner, M.; Mayr, J.; Jesina, P.; Vesela, K.; Wenchich, L.; Szentivanyi, K.; Hansikova, H.; Sperl, W.; et al. Neonatal Onset of Mitochondrial Disorders in 129 Patients: Clinical and Laboratory Characteristics and a New Approach to Diagnosis. J. Inherit. Metab. Dis. 2012, 35, 749–759.
- Kenvin, S.; Torregrosa-Muñumer, R.; Reidelbach, M.; Pennonen, J.; Turkia, J.J.; Rannila, E.; Kvist, J.; Sainio, M.T.; Huber, N.; Herukka, S.-K.; et al. Threshold of Heteroplasmic Truncating MT-ATP6 Mutation in Reprogramming, Notch Hyperactivation and Motor Neuron Metabolism. Hum. Mol. Genet. 2022, 31, 958–974.
- Kucharczyk, R.; Salin, B.; Di Rago, J.-P. Introducing the Human Leigh Syndrome Mutation T9176G into Saccharomyces Cerevisiae Mitochondrial DNA Leads to Severe Defects in the Incorporation of Atp6p into the ATP Synthase and in the Mitochondrial Morphology. Hum. Mol. Genet. 2009, 18, 2889–2898.
- Carrozzo, R.; Tessa, A.; Vazquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schagger, H.; et al. The T9176G mtDNA Mutation Severely Affects ATP Production and Results in Leigh Syndrome. Neurology 2001, 56, 687–690.
- Kucharczyk, R.; Ezkurdia, N.; Couplan, E.; Procaccio, V.; Ackerman, S.H.; Blondel, M.; Di Rago, J.-P. Consequences of the Pathogenic T9176C Mutation of Human Mitochondrial DNA on Yeast Mitochondrial ATP Synthase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 1105–1112.
- Thyagarajan, D.; Shanske, S.; Vazquez -Memije, M.; Devivo, D.; Dimauro, S. A Novel Mitochondrial ATPase 6 Point Mutation in Familial Bilateral Striatal Necrosis. Ann. Neurol. 1995, 38, 468–472.
- Verny, C.; Guegen, N.; Desquiret, V.; Chevrollier, A.; Prundean, A.; Dubas, F.; Cassereau, J.; Ferre, M.; Amati-Bonneau, P.; Bonneau, D.; et al. Hereditary Spastic Paraplegia-like Disorder Due to a Mitochondrial ATP6 Gene Point Mutation. Mitochondrion 2011, 11, 70–75.
- Kabala, A.M.; Lasserre, J.-P.; Ackerman, S.H.; Di Rago, J.-P.; Kucharczyk, R. Defining the Impact on Yeast ATP Synthase of Two Pathogenic Human Mitochondrial DNA Mutations, T9185C and T9191C. Biochimie 2014, 100, 200–206.
- Moslemi, A.-R.; Darin, N.; Tulinius, M.; Oldfors, A.; Holme, E. Two New Mutations in the MTATP6 Gene Associated with Leigh Syndrome. Neuropediatrics 2005, 36, 314–318.
- Pitceathly, R.D.S.; Murphy, S.M.; Cottenie, E.; Chalasani, A.; Sweeney, M.G.; Woodward, C.; Mudanohwo, E.E.; Hargreaves, I.; Heales, S.; Land, J.; et al. Genetic Dysfunction of MT-ATP6 Causes Axonal Charcot-Marie-Tooth Disease. Neurology 2012, 79, 1145–1154.
- Castagna, A.E.; Addis, J.; McInnes, R.R.; Clarke, J.T.R.; Ashby, P.; Blaser, S.; Robinson, B.H. Late Onset Leigh Syndrome and Ataxia Due to a T to C Mutation at Bp 9,185 of Mitochondrial DNA. Am. J. Med. Genet. 2007, 143A, 808–816.
- Saneto, R.P.; Singh, K.K. Illness-Induced Exacerbation of Leigh Syndrome in a Patient with the MTATP6 Mutation, m. 9185 T>C. Mitochondrion 2010, 10, 567–572.
- Pfeffer, G.; Blakely, E.L.; Alston, C.L.; Hassani, A.; Boggild, M.; Horvath, R.; Samuels, D.C.; Taylor, R.W.; Chinnery, P.F. Adult-Onset Spinocerebellar Ataxia Syndromes Due to MTATP6 Mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 883–886.
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674.e9.
- Kucharczyk, R.; Dautant, A.; Gombeau, K.; Godard, F.; Tribouillard-Tanvier, D.; Di Rago, J.-P. The Pathogenic MT-ATP6 m.8851T>C Mutation Prevents Proton Movements within the n-Side Hydrophilic Cleft of the Membrane Domain of ATP Synthase. Biochim. Biophys. Acta (BBA)-Bioenerg. 2019, 1860, 562–572.
- Ješina, P.; Tesařová, M.; Fornůsková, D.; Vojtíšková, A.; Pecina, P.; Kaplanová, V.; Hansíková, H.; Zeman, J.; Houštěk, J. Diminished Synthesis of Subunit a (ATP6) and Altered Function of ATP Synthase and Cytochrome c Oxidase Due to the mtDNA 2 Bp Microdeletion of TA at Positions 9205 and 9206. Biochem. J. 2004, 383, 561–571.
- Seneca, S.; Abramowicz, M.; Lissens, W.; Muller, M.F.; Vamos, E.; De Meirleir, L. A Mitochondrial DNA Microdeletion in a Newborn Girl with Transient Lactic Acidosis. J. Inherit. Metab. Dis. 1996, 19, 115–118.
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; Di Rago, J.-P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329.
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225.
- Ganetzky, R.D.; Stendel, C.; McCormick, E.M.; Zolkipli-Cunningham, Z.; Goldstein, A.C.; Klopstock, T.; Falk, M.J. MT-ATP6 Mitochondrial Disease Variants: Phenotypic and Biochemical Features Analysis in 218 Published Cases and Cohort of 14 New Cases. Hum. Mutat. 2019, 40, 499–515.
- Morava, E.; Rodenburg, R.J.; Hol, F.; De Vries, M.; Janssen, A.; Van Den Heuvel, L.; Nijtmans, L.; Smeitink, J. Clinical and Biochemical Characteristics in Patients with a High Mutant Load of the Mitochondrial T8993G/C Mutations. Am. J. Med. Genet. 2006, 140A, 863–868.
- Na, J.; Lee, Y. Genotype-phenotype Analysis of MT-ATP6 -associated Leigh Syndrome. Acta Neuro Scand. 2022, 145, 414–422.
- Solaini, G.; Sgarbi, G.; Lenaz, G.; Baracca, A. Evaluating Mitochondrial Membrane Potential in Cells. Biosci. Rep. 2007, 27, 11–21.
- Rieger, B.; Arroum, T.; Borowski, M.-T.; Villalta, J.; Busch, K.B. Mitochondrial F1FO ATP Synthase Determines the Local Proton Motive Force at Cristae Rims. EMBO Rep. 2021, 22, e52727.
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Interface Mobility between Monomers in Dimeric Bovine ATP Synthase Participates in the Ultrastructure of Inner Mitochondrial Membranes. Proc. Natl. Acad. Sci. USA 2021, 118, e2021012118.
- Bénit, P.; El-Khoury, R.; Schiff, M.; Sainsard-Chanet, A.; Rustin, P. Genetic Background Influences Mitochondrial Function: Modeling Mitochondrial Disease for Therapeutic Development. Trends Mol. Med. 2010, 16, 210–217.
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic Hybrid (Cybrid) Cell Lines as a Practical Model for Mitochondriopathies. Redox Biol. 2014, 2, 619–631.
- McKnight, C.L.; Low, Y.C.; Elliott, D.A.; Thorburn, D.R.; Frazier, A.E. Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned? IJMS 2021, 22, 7730.
- Palombo, F.; Peron, C.; Caporali, L.; Iannielli, A.; Maresca, A.; Di Meo, I.; Fiorini, C.; Segnali, A.; Sciacca, F.L.; Rizzo, A.; et al. The Relevance of Mitochondrial DNA Variants Fluctuation during Reprogramming and Neuronal Differentiation of Human iPSCs. Stem Cell Rep. 2021, 16, 1953–1967.
- Lorenz, C.; Zink, A.; Henke, M.-T.; Staege, S.; Mlody, B.; Bünning, M.; Wanker, E.; Diecke, S.; Schuelke, M.; Prigione, A. Generation of Four iPSC Lines from Four Patients with Leigh Syndrome Carrying Homoplasmic Mutations m.8993T > G or m.8993T > C in the Mitochondrial Gene MT-ATP6. Stem Cell Res. 2022, 61, 102742.
- Galera-Monge, T.; Zurita-Díaz, F.; Moreno-Izquierdo, A.; Fraga, M.F.; Fernández, A.F.; Ayuso, C.; Garesse, R.; Gallardo, M.E. Generation of a Human iPSC Line from a Patient with an Optic Atrophy “plus” Phenotype Due to a Mutation in the OPA1 Gene. Stem Cell Res. 2016, 16, 673–676.
- Grace, H.E.; Galdun, P.; Lesnefsky, E.J.; West, F.D.; Iyer, S. mRNA Reprogramming of T8993G Leigh’s Syndrome Fibroblast Cells to Create Induced Pluripotent Stem Cell Models for Mitochondrial Disorders. Stem Cells Dev. 2019, 28, 846–859.
- Henke, M.-T.; Zink, A.; Diecke, S.; Prigione, A.; Schuelke, M. Generation of Two Mother–Child Pairs of iPSCs from Maternally Inherited Leigh Syndrome Patients with m.8993 T > G and m.9176 T > G MT-ATP6 Mutations. Stem Cell Res. 2023, 67, 103030.
- Lai, Y.; Zhang, Y.; Zhou, S.; Xu, J.; Du, Z.; Feng, Z.; Yu, L.; Zhao, Z.; Wang, W.; Tang, Y.; et al. Structure of the Human ATP Synthase. Mol. Cell 2023, 83, 2137–2147.e4.
- Steiner, T.; Zink, A.; Henke, M.-T.; Cecchetto, G.; Buenning, M.; Rossi, A.; Schuelke, M.; Prigione, A. RNA-Based Generation of iPSCs from a Boy Carrying the Mutation m.9185 T>C in the Mitochondrial Gene MT-ATP6 and from His Healthy Mother. Stem Cell Res. 2022, 64, 102920.
- Licchetta, L.; Ferri, L.; La Morgia, C.; Zenesini, C.; Caporali, L.; Lucia Valentino, M.; Minardi, R.; Fulitano, D.; Di Vito, L.; Mostacci, B.; et al. Epilepsy in MT - ATP6 - Related Mils/NARP: Correlation of Elettroclinical Features with Heteroplasmy. Ann. Clin. Transl. Neurol. 2021, 8, 704–710.
- Angural, A.; Sharma, I.; Pandoh, P.; Sharma, V.; Spolia, A.; Rai, E.; Singh, V.; Razdan, S.; Pandita, K.K.; Sharma, S. A Case Report on a Novel MT-ATP6 Gene Variation in Atypical Mitochondrial Leigh Syndrome Associated with Bilateral Basal Ganglia Calcifications. Mitochondrion 2019, 46, 209–213.
- Lehmann Urban, D.; Motlagh Scholle, L.; Wagner, M.; Ludolph, A.C.; Rosenbohm, A. The m.9143T>C Variant: Recurrent Infections and Immunodeficiency as an Extension of the Phenotypic Spectrum in MT-ATP6 Mutations? Diseases 2020, 8, 19.
- Sainio, M.T.; Aaltio, J.; Hyttinen, V.; Kortelainen, M.; Ojanen, S.; Paetau, A.; Tienari, P.; Ylikallio, E.; Auranen, M.; Tyynismaa, H. Effectiveness of Clinical Exome Sequencing in Adult Patients with Difficult-to-diagnose Neurological Disorders. Acta Neuro Scand. 2022, 145, 63–72.
- Birtel, J.; Von Landenberg, C.; Gliem, M.; Gliem, C.; Reimann, J.; Kunz, W.S.; Herrmann, P.; Betz, C.; Caswell, R.; Nesbitt, V.; et al. Mitochondrial Retinopathy. Ophthalmol. Retin. 2022, 6, 65–79.
- Nolte, D.; Kang, J.-S.; Hofmann, A.; Schwaab, E.; Krämer, H.H.; Müller, U. Mutations in MT-ATP6 Are a Frequent Cause of Adult-Onset Spinocerebellar Ataxia. J. Neurol. 2021, 268, 4866–4873.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
591
Revisions:
2 times
(View History)
Update Date:
01 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No