 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Ungureanu | -- | 5277 | 2023-09-12 06:16:22 | | | |
| 2 | Peter Tang | Meta information modification | 5277 | 2023-09-12 07:24:13 | | |
Video Upload Options
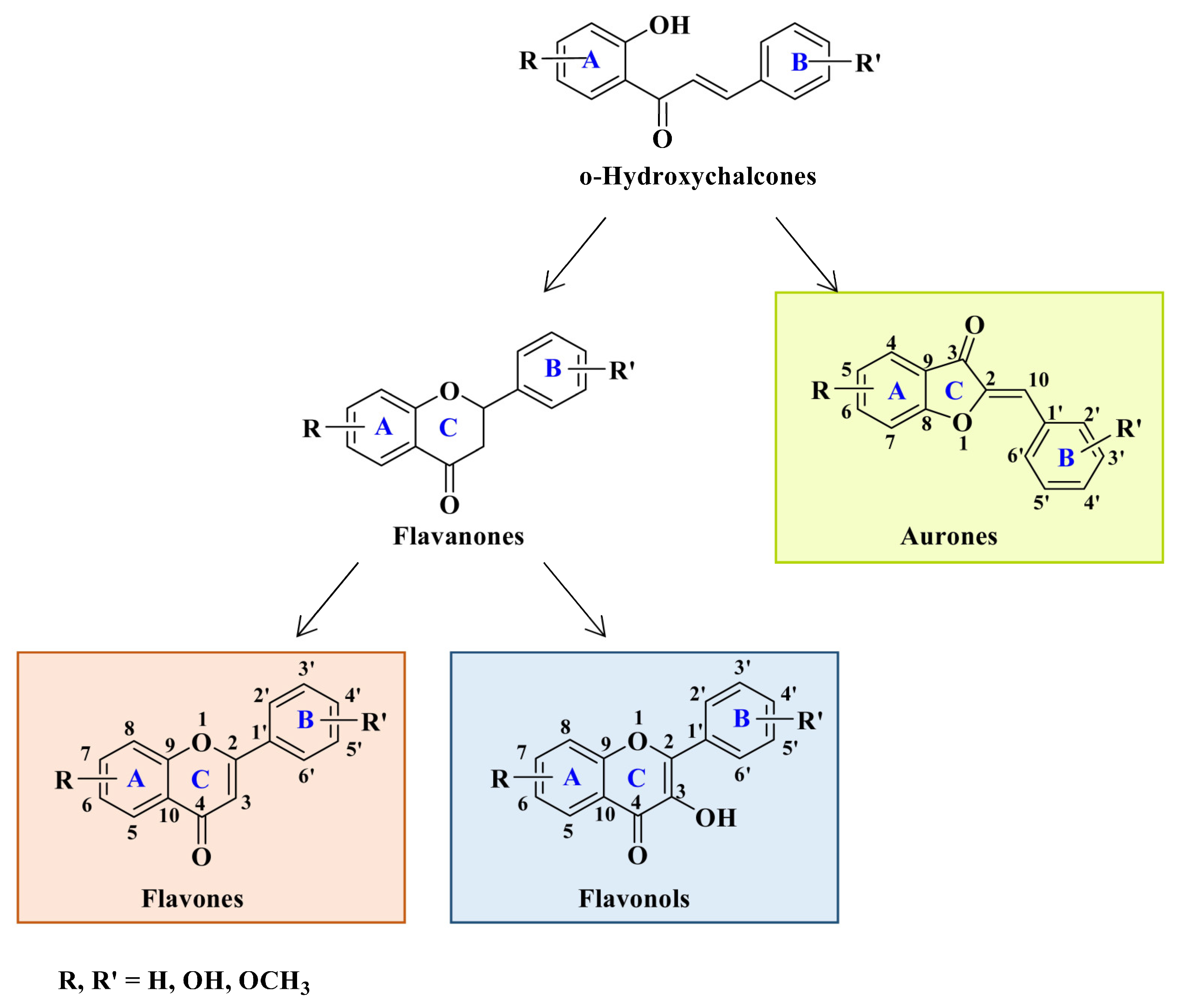
Flavonoids are a widely distributed group of natural polyphenolic compounds that are found in plants usually in glycosylated form and have been shown to possess a wide range of biological activities, including antioxidant, anti-inflammatory, antibacterial, antiviral, and anticancer properties, making them an attractive target for synthesis and further research.
1. Introduction
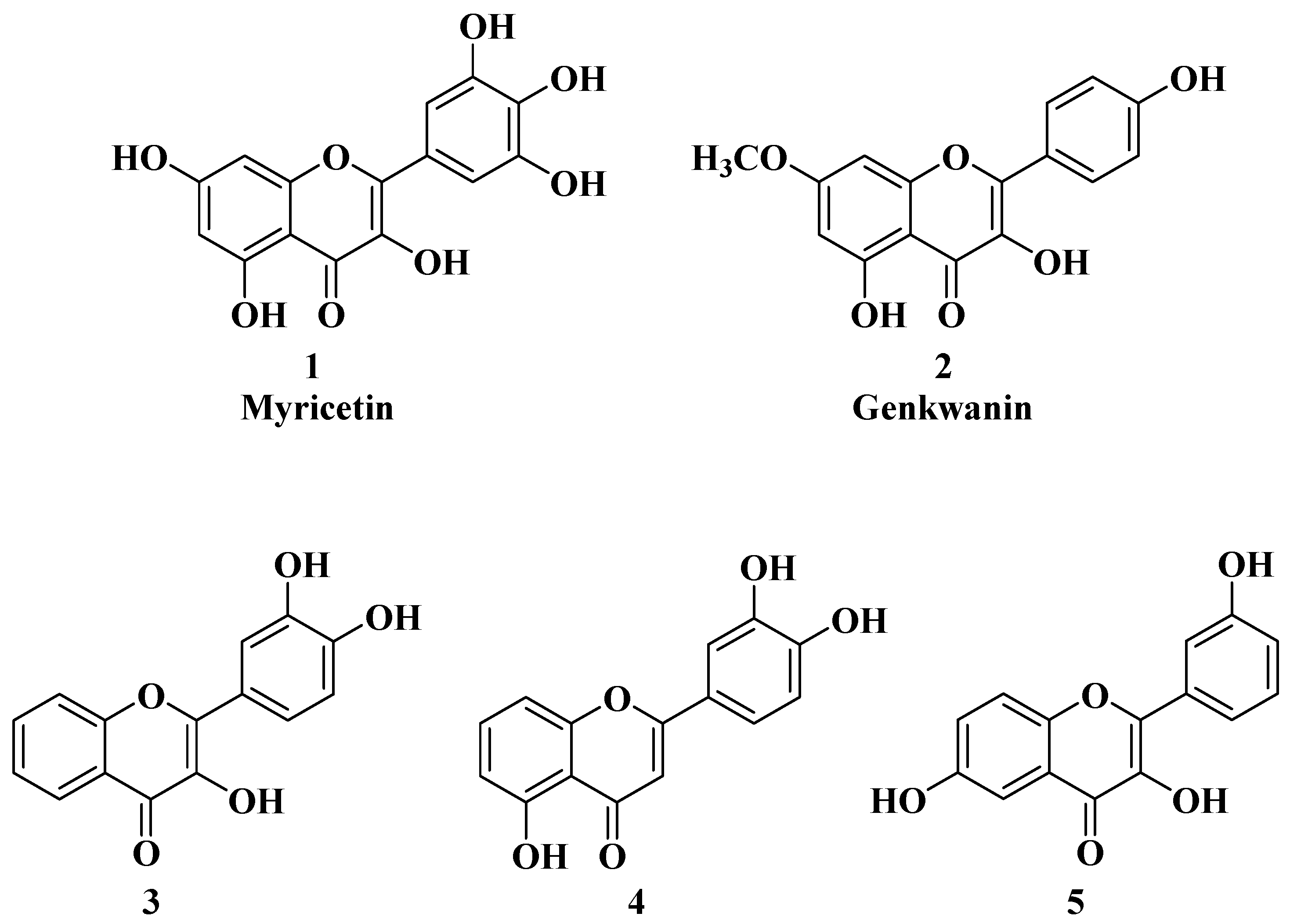
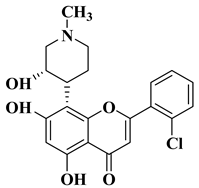
2. Anticancer Activity
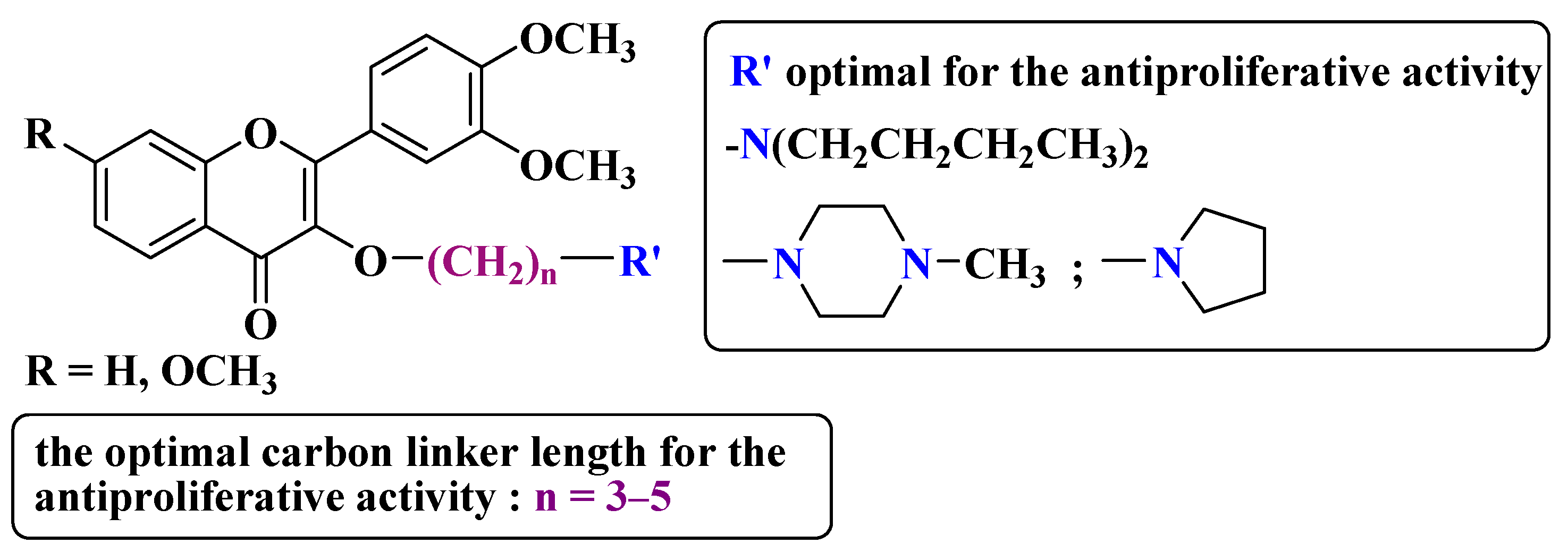
|
Entry |
Chemical Structure |
Cancer Cell Lines against the Tested Compounds Present Cytotoxic Activity |
Ref. |
|---|---|---|---|
|
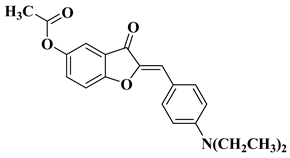
1 |
|
|
[42] |
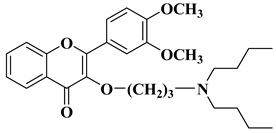
|
2 |
|
R = OCH3
|
[28] |
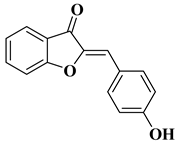
|
R = OH
|
|||
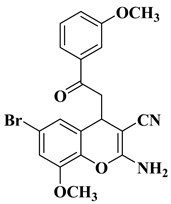
|
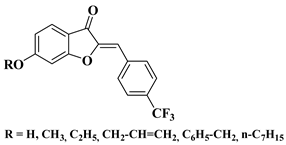
3 |
|
|
[28] |
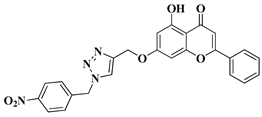
|
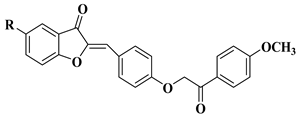
4 |
|
|
[28] |
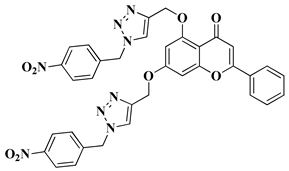
|
5 |
|
melanoma (160.26–107.81% SKMEL5), hematologic (111.12–92.74% leukemia HL60), renal (129.05% RXF393), colon (98.27–82.03% COLO205), lung (93.28% H522), brain (147.04–141.63% SF295 glioma), ovarian (76.54–51.79% IGROV1, OVCAR3, OVCAR8, ADREES, SKOV3), growth inhibition determined at 10 µM dosage. |
[29] |
|
6 |
|
|
[30] |
|
7 |
|
|
[8] |
|
8 |
|
|
[31] |
|
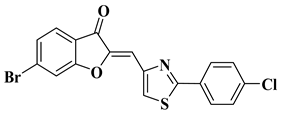
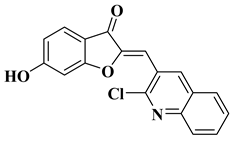
9 |
|
CNS cancer: SF-268 (GI50 = 3.52 µM), SF-295 (GI50 = 2.32 µM), SF-539 (GI50 = 2.21 µM), SNB-19 (GI50 = 4.55 µM), SNB-75 (GI50 = 1.69 µM), U251 (GI50 = 2.80 µM).
|
[31] |
|
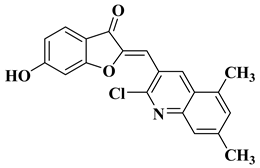
10 |
|
|
[32] |
|
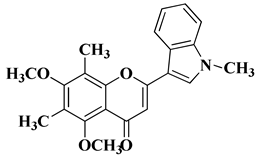
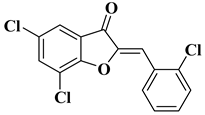
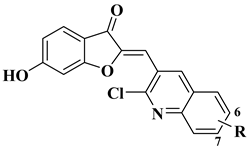
11 |
|
|
[33] |
|
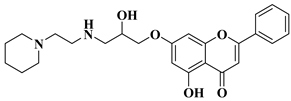
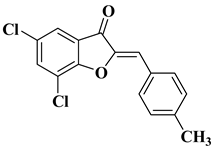
12 |
|
|
[33] |
|
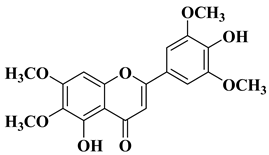
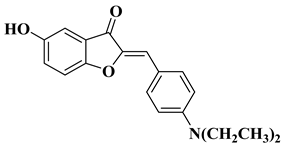
13 |
|
|
[33] |
|
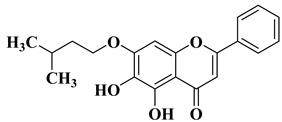
14 |
|
|
[33] |
|
15 |
|
|
[43] |
|
16 |
|
|
[44] |
|
17 |
|
|
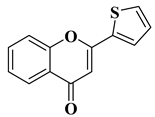
[9] |
|
18 |
|
|
[45] |
|
19 |
|
|
[45] |
|
20 |
|
|
[45] |
|
21 |
|
|
[41] |
|
22 |
|
|
[41] |
|
23 |
|
|
[46] |
|
24 |
|
|
[46] |
|
25 |
|
|
[47] |
|
26 |
|
|
[47] |
|
27 |
|
|
[48] |
|
28 |
|
|
[49] |
|
29 |
|
R = Cl: leukemia cell lines MOLT-4 (−17.79% mean growth percentage), and SR (−22.38% mean growth percentage). R = H: renal cancer cell line UO-31 (−44.36% mean growth percentage). The mean growth percentages were determined for five concentrations ranging from 10−4 to 10−8 M. |
[50] |
|
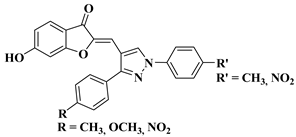
30 |
|
R′ = CH3 and R = CH3, OCH3, NO2 (IC50 = 25–28.3 µM). R′ = NO2 and R = NO2 (IC50 = 25.1 µM). |
[51] |
|
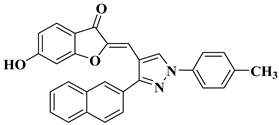
31 |
|
|
[51] |
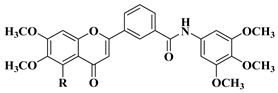
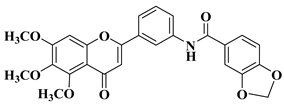
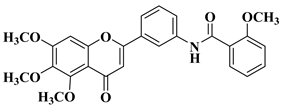
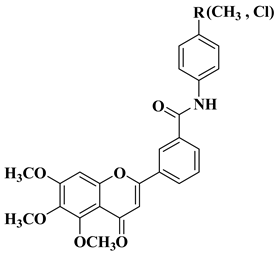
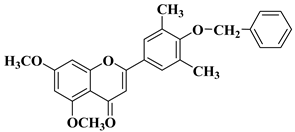
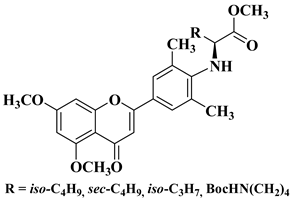
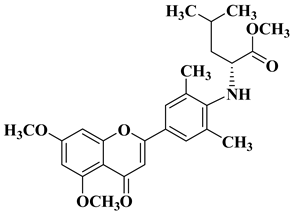
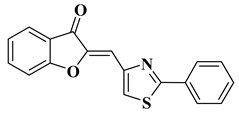
3. Antibacterial and Antifungal Activity
4. Antiviral Activity
|
Entry |
Chemical Structure |
Microbial Strains against the Tested Compounds Present Antimicrobial Activity |
Ref. |
|---|---|---|---|
|
1 |
|
Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 16 mm) Penicillium italicum (IZ = 20 mm) Fusarium oxysporum (IZ = 31 mm) |
[53] |
|
2 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 31 mm) Pseudomonas aeruginosa (IZ = 11 mm) Escherichia coli (IZ = 30 mm) |
[53] |
|
3 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 30 mm) Bacillus subtilis (IZ = 11 mm) Escherichia coli (IZ = 31 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 13 mm) Penicillium italicum (IZ = 24 mm) Fusarium oxysporum (IZ = 25 mm) |
[53] |
|
4 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 33 mm) Bacillus subtilis (IZ = 17 mm) Escherichia coli (IZ = 33 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 14 mm) Penicillium italicum (IZ = 26 mm) Fusarium oxysporum (IZ = 27 mm) |
[53] |
|
5 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 2 mg/L) Escherichia coli (MIC = 4 mg/L) Salmonella gallinarum (MIC = 0.125 mg/L) |
[54] |
|
6 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 1 mg/L) Escherichia coli (MIC = 2 mg/L) Salmonella gallinarum (MIC = 0.05 mg/L) Listeria monocytogenes (MIC = 0.5 mg/L) |
[54] |
|
7 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (81.33%; 100%) Penicillium expansum (60.87%; 100%) Aspergillus flavus (41.02%; 65.64%) |
[55] |
|
8 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (70%; 100%) Penicillium expansum (42.15%; 100%) Aspergillus flavus (6.41%; 46.15%) |
[55] |
|
9 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (76.88%; 100%) Aspergillus flavus (15.38%; 60.51%) |
[55] |
|
10 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (73.33%; 100%) |
[55] |
|
11 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 0.02 mg/mL) Mycobacterium smegmatis (MIC = 0.625 mg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.625 mg/mL) |
[56] |
|
12 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 2.5 mg/mL) Bacillus subtilis (MIC = 0.156 mg/mL) Mycobacterium smegmatis (MIC = 0.078 mg/mL) Anti biofilm and anti quorum sensing activity (100 μg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.313 mg/mL) Candida albicans (MIC = 0.078 mg/mL) |
[56] |
|
13 |
|
Antibacterial activity (R=6-OCH3, 7-Cl) Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 1.25 mg/mL) Klebsiella pneumoniae (MIC = 0.625 mg/mL) Anti biofilm activity (R=6-OCH3, 100 μg/mL) Antifungal activity (R=7-Cl): Candida albicans (MIC = 0.156 mg/mL) |
[56] |
|
14 |
|
Antibacterial activity Mycobacterium tuberculosis H37Rv (MIC = 6.25 µg/mL) |
[33] |
|
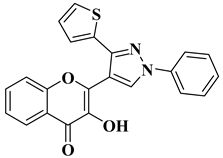
15 |
|
Antiviral activity Human cytomegalovirus (EC50 = 0.126 nM) |
[58] |
|
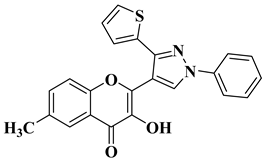
16 |
|
Antiviral activity Chikungunya Virus (IC50 = 0.44 µM) |
[59] |
|
17 |
|
Antiviral activity Chikungunya Virus (IC50 = 0.45 µM) |
[59] |
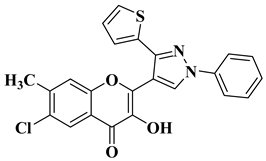
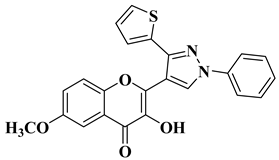
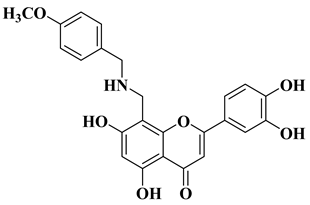
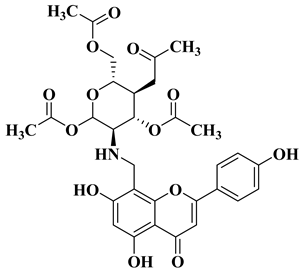
References
- Berim, A.; Gang, D.R. Methoxylated flavones: Occurrence, importance, biosynthesis. Phytochem. Rev. 2016, 15, 363–390.
- Ververidis, F.; Trantas, E.; Douglas, C.; Vollmer, G.; Kretzschmar, G.; Panopoulos, N. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2007, 2, 1214–1234.
- Hou, D.X.; Kumamoto, T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: Direct binding and molecular modeling. Antioxid. Redox Signal. 2010, 13, 691–719.
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011, 2, e182.
- Golub, A.G.; Bdzhola, V.G.; Ostrynska, O.V.; Kyshenia, I.V.; Sapelkin, V.M.; Prykhod’ko, A.O.; Kukharenko, O.P.; Yarmoluk, S.M. Discovery and characterization of synthetic 4′-hydroxyflavones—New CK2 inhibitors from flavone family. Bioorg. Med. Chem. 2013, 21, 6681–6689.
- Chao, S.W.; Su, M.Y.; Chiou, L.C.; Chen, L.C.; Chang, C.I.; Huang, W.J. Total Synthesis of Hispidulin and the Structural Basis for Its Inhibition of Proto-oncogene Kinase Pim-1. J. Nat. Prod. 2015, 78, 1969–1976.
- Sedlacek, H.; Czech, J.; Naik, R.; Kaur, G.; Worland, P.; Losiewicz, M.; Parker, B.; Carlson, B.; Smith, A.; Senderowicz, A.; et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996, 9, 1143–1168.
- Pontes, O.; Costa, M.; Santos, F.; Sampaio-Marques, B.; Dias, T.; Ludovico, P.; Baltazar, F.; Proença, F. Exploitation of new chalcones and 4H-chromenes as agents for cancer treatment. Eur. J. Med. Chem. 2018, 157, 101–114.
- Monasterio, A.; Urdaci, M.; Pinchuk, I.; Moratalla, N.; Martínez, I. Flavonoids induce apoptosis in human leukemia U937 cells through caspase-and caspase-calpain-dependent pathways. Nutr. Cancer 2004, 50, 90–100.
- Moreira, J.; Ribeiro, D.; Silva, P.M.A.; Nazareth, N.; Monteiro, M.; Palmeira, A.; Saraiva, L.; Pinto, M.; Bousbaa, H.; Cidade, H. New alkoxy flavone derivatives targeting caspases: Synthesis and antitumor activity evaluation. Molecules 2019, 24, 129.
- Kariagina, A.; Doseff, A.I. Anti-Inflammatory Mechanisms of Dietary Flavones: Tapping into Nature to Control Chronic Inflammation in Obesity and Cancer. Int. J. Mol. Sci. 2022, 23, 15753.
- Occhiuto, C.J.; Moerland, J.A.; Leal, A.S.; Gallo, K.A.; Liby, K.T. The Multi-Faceted Consequences of NRF2 Activation throughout Carcinogenesis. Mol. Cells 2023, 46, 176–186.
- Ross, J.A.; Kasum, C.M. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 2002, 22, 19–34.
- Jiang, M.; Zhu, M.; Wang, L.; Yu, S. Anti-tumor effects and associated molecular mechanisms of myricetin. Biomed. Pharmacother. 2019, 120, 109506.
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A dietary molecule with diverse biological activities. Nutrients 2016, 8, 90.
- Zang, W.; Wang, T.; Wang, Y.; Li, M.; Xuan, X.; Ma, Y.; Zhao, G. Myricetin exerts anti-proliferative, anti-invasive, and pro-apoptotic effects on esophageal carcinoma EC9706 and KYSE30 cells via RSK2. Tumor Biol. 2014, 35, 12583–12592.
- Chen, Y.C.; He, X.L.; Qi, L.; Shi, W.; Yuan, L.-W.; Huang, M.-Y.; Xu, Y.-L.; Chen, X.; Gu, L.; Zhang, L.-L.; et al. Myricetin inhibits interferon-γ-induced PD-L1 and IDO1 expression in lung cancer cells. Biochem. Pharmacol. 2022, 197, 114940.
- Han, S.-H.; Lee, J.-H.; Woo, J.-S.; Jung, G.-H.; Jung, S.-H.; Han, E.-J.; Kim, B.; Cho, S.-D.; Nam, J.-S.; Che, J.H.; et al. Myricetin induces apoptosis and autophagy in human gastric cancer cells through inhibition of the PI3K/Akt/mTOR pathway. Heliyon 2022, 8, e09309.
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273.
- Li, L.; Ma, H.; Li, D.; Shu, Q.; Wang, T.; Song, X.; Xu, H. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through inhibition of HIF-1α. Toxicology and Appl. Pharmacol. 2022, 454, 116246.
- Gu, L.; Li, Z.; Zhang, X.; Chen, M.; Zhang, X. Identification of MAP Kinase Kinase 3 as a protein target of myricetin in non-small cell lung cancer cells. Biomed. Pharmacother. 2023, 161, 114460.
- El Menyiy, N.; Aboulaghras, S.; Bakrim, S.; Moubachir, R.; Taha, D.; Khalid, A.; Abdalla, A.N.; Algarni, A.S.; Hermansyah, A.; Ming, L.C.; et al. Genkwanin: An emerging natural compound with multifaceted pharmacological effects. Biomed. Pharmacother. 2023, 165, 115159.
- Li, Y.; Hong, J.; Li, H.; Qi, X.; Guo, Y.; Han, M.; Wang, X. Genkwanin nanosuspensions: A novel and potential antitumor drug in breast carcinoma therapy. Drug Deliv. 2017, 24, 1491–1500.
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants 2020, 9, 461.
- Grigalius, I.; Petrikaite, V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules 2017, 22, 2169.
- Zhao, L.; Yuan, X.; Wang, J.; Feng, Y.; Ji, F.; Li, Z.; Bian, J. A review on flavones targeting serine/threonine protein kinases for potential anticancer drugs. Bioorg. Med. Chem. 2019, 27, 677–685.
- Chao, S.H.; Price, D.H. Flavopiridol Inactivates P-TEFb and Blocks Most RNA Polymerase II Transcription in Vivo. J. Biol. Chem. 2001, 276, 31793–31799.
- Hassan, A.H.; Choi, E.; Yoon, Y.M.; Lee, K.W.; Yoo, S.Y.; Cho, M.C.; Yang, J.S.; Kim, H.I.; Hong, J.Y.; Shin, J.-S.; et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur. J. Med. Chem. 2019, 161, 559–580.
- Hassan, A.H.E.; Lee, K.T.; Lee, Y.S. Flavone-based arylamides as potential anticancers: Design, synthesis and in vitro cell-based/cell-free evaluations. Eur. J. Med. Chem. 2020, 187, 111965.
- Li, X.; Zhang, C.; Guo, S.; Rajaram, P.; Lee, M.; Chen, G.; Fong, R.; Gonzalez, A.; Zhang, Q.; Zheng, S.; et al. Structure-activity relationship and pharmacokinetic studies of 3-O-substitutedflavonols as anti-prostate cancer agents. Eur. J. Med. Chem. 2018, 157, 978–993.
- Németh-Rieder, A.; Keglevich, P.; Hunyadi, A.; Latif, A.D.; Zupkó, I.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Flavone—1,2,3-Triazole Hybrids. Molecules 2023, 28, 626.
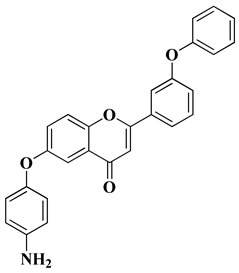
- Mobbili, G.; Romaldi, B.; Sabbatini, G.; Amici, A.; Marcaccio, M.; Galeazzi, R.; Laudadio, E.; Armeni, T.; Minnelli, C. Identification of Flavone Derivative Displaying a 4-Aminophenoxy Moiety as Potential Selective Anticancer Agent in NSCLC Tumor Cells. Molecules 2023, 28, 3239.
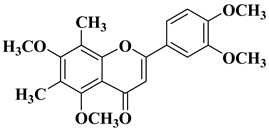
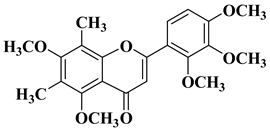
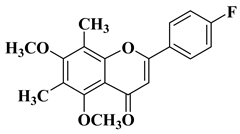
- Bollikolla, H.B.; Anandam, R.; Chinnam, S.; Varala, R.; Khandapu, B.M.K.; Kapavarapu, R.; Syed, K.S.; Dubasi, N.; Syed, M.A. C-Dimethylated Flavones as Possible Potential Anti-Tubercular and Anticancer Agents. Chem. Biodivers. 2023, 20, e202201201.
- Schoepfer, J.; Fretz, H.; Chaudhuri, B.; Muller, L.; Seeber, E.; Meijer, L.; Lozach, O.; Vangrevelinghe, E.; Furet, P. Structure-based design and synthesis of 2-benzylidene-benzofuran-3-ones as flavopiridol mimics. J. Med. Chem. 2002, 45, 1741–1747.
- Priyadarshani, G.; Nayak, A.; Amrutkar, S.M.; Das, S.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C. Scaffold-Hopping of Aurones: 2-Arylideneimidazopyridinones as Topoisomerase IIα-Inhibiting Anticancer Agents. ACS Med. Chem. Lett. 2016, 7, 1056–1061.
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969.
- Lawrence, N.J.; Rennison, D.; McGown, A.T.; Hadfield, J.A. The total synthesis of an aurone isolated from Uvaria hamiltonii: Aurones and flavones as anticancer agents. Bioorg. Med. Chem. Lett. 2003, 13, 3759–3763.
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg. Med. Chem. 2009, 17, 8073–8085.
- Hadjeri, M.; Barbier, M.; Ronot, X.; Mariotte, A.M.; Boumendjel, A.; Boutonnat, J. Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J. Med. Chem. 2003, 46, 2125–2131.
- Sim, H.M.; Wu, C.P.; Ambudkar, S.V.; Go, M.L. In vitro and in vivo modulation of ABCG2 by functionalized aurones and structurally related analogs. Biochem. Pharmacol. 2011, 82, 1562–1571.
- Coman, F.M.; Mbaveng, A.T.; Marc, G.; Leonte, D.; Brém, B.; Vlase, L.; Imre, S.; Kuete, V.; Zaharia, V. Heterocycles 47. Synthesis, Characterization and Biological Evaluation of some New Thiazole Aurones as Antiproliferative Agents. Farmacia 2020, 68, 492–506.
- Semenov, I.; Akyuz, C.; Roginskaya, V.; Chauhan, D.; Corey, S.J. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk. Res. 2002, 26, 271–280.
- Jeon, K.-H.; Park, S.; Shin, J.-H.; Jung, A.-R.; Hwang, S.-Y.; Seo, S.H.; Jo, H.; Na, Y.; Kwon, Y. Synthesis and evaluation of 7-(3-aminopropyloxy)-substituted flavone analogue as a topoisomerase IIα catalytic inhibitor and its sensitizing effect to enzalutamide in castration-resistant prostate cancer cells. Eur. J. Med. Chem. 2023, 246, 114999.
- Su, L.; Li, W.; Liu, K.; Wang, Q. Synthesis and anti-proliferative activities of 5,6,7-trimethoxyflavones and their derivatives. Nat. Prod. Res. 2022, 36, 4070–4075.
- Zhang, N.; Yang, J.; Li, K.; Luo, J.; Yang, S.; Song, J.-R.; Chen, C.; Pan, W.-D. Synthesis of flavone derivatives via N-amination and evaluation of their anticancer activities. Molecules 2019, 24, 2723.
- Elhadi, A.A.; Osman, H.; Iqbal, M.A.; Rajeswari, S.K.; Ahamed, M.B.K.; Majid, A.M.A.; Rosli, M.M.; Razak, I.A.; Majid, A.S.A. Synthesis and structural elucidation of two new series of aurone derivatives as potent inhibitors against the proliferation of human cancer cells. Med. Chem. Res. 2015, 24, 3504–3515.
- Cheng, H.; Zhang, L.; Liu, Y.; Chen, S.; Cheng, H.; Lu, X.; Zheng, Z.; Zhou, G.-C. Design, synthesis and discovery of 5-hydroxyaurone derivatives as growth inhibitors against HUVEC and some cancer cell lines. Eur. J. Med. Chem. 2010, 45, 5950–5957.
- Zheng, X.; Wang, H.; Liu, Y.-M.; Yao, X.; Tong, M.; Wang, Y.-H.; Liao, D.-F. Synthesis, Characterization, and Anticancer Effect of Trifluoromethylated Aurone Derivatives. J. Heterocycl. Chem. 2014, 6, 1098–1107.
- Uesawa, Y.; Sakagami, H.; Ikezoe, N.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure-cytotoxicity relationship of aurones. Anticancer Res. 2017, 37, 6169–6176.
- Demirayak, S.; Yurttas, L.; Gundogdu-Karaburun, N.; Karaburun, A.C.; Kayagil, I. Synthesis and anticancer activity evaluation of new aurone derivatives. J. Enzym. Inhib. Med. Chem. 2015, 30, 816–825.
- Lathwal, E.; Kumar, S.; Sahoo, P.K.; Ghosh, S.; Mahata, S.; Nasare, V.D.; Kumar, S. Synthesis, cytotoxic evaluation and structure activity relationship of pyrazole hybrid aurones on gastric cancer (AGS) cell lines. Results Chem. 2022, 4, 100590.
- Gao, L.; Tang, Z.; Li, T.; Wang, J. Myricetin exerts anti-biofilm activity and attenuates osteomyelitis by inhibiting the TLR2/MAPK pathway in experimental mice. Microb. Pathog. 2023, 182, 106165.
- Ashok, D.; Kifah, M.A.; Lakshmi, B.V.; Sarasija, M.; Adam, S. Microwave-assisted one-pot synthesis of some new flavonols by modified Algar–Flynn–Oyamada reaction and their antimicrobial activity. Chem. Heterocycl. Compd. 2016, 52, 172–176.
- Lv, X.H.; Liu, H.; Ren, Z.L.; Wang, W.; Tang, F.; Cao, H.Q. Design, synthesis and biological evaluation of novel flavone Mannich base derivatives as potential antibacterial agents. Mol. Divers. 2019, 23, 299–306.
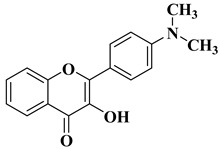
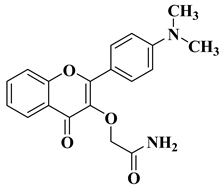
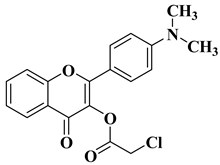
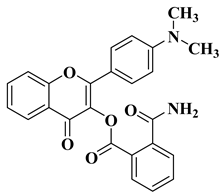
- Khdera, H.A.; Saad, S.Y.; Moustapha, A.; Kandil, F. Synthesis of new flavonoid derivatives based on 3-hydroxy-4′-dimethylamino flavone and study the activity of some of them as antifungal. Heliyon 2022, 8, e12062.
- Kumar, G.; Lathwal, E.; Saroha, B.; Kumar, S.; Kumar, S.; Chauhan, N.S.; Kumar, T. Synthesis and Biological Evaluation of Quinoline-Based Novel Aurones. ChemistrySelect 2020, 5, 3539–3543.
- Pan, H.; He, J.; Yang, Z.; Yao, X.; Zhang, H.; Li, R.; Xiao, Y.; Zhao, C.; Jiang, H.; Liu, Y.; et al. Myricetin possesses the potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation in rats and mice. Phytomedicine 2023, 116, 154858.
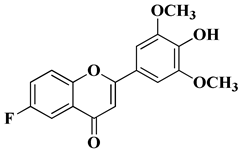
- Fujimoto, K.J.; Nema, D.; Ninomiya, M.; Koketsu, M.; Sadanari, H.; Takemoto, M.; Daikoku, T.; Murayama, T. An in silico-designed flavone derivative, 6-fluoro-4′-hydroxy-3′,5′-dimetoxyflavone, has a greater anti-human cytomegalovirus effect than ganciclovir in infected cells. Antivir. Res. 2018, 154, 10–16.
- Badavath, V.N.; Jadav, S.; Pastorino, B.; de Lamballerie, X.; Sinha, N.B.; Jayaprakash, V. Synthesis and Antiviral Activity of 2-aryl-4H-chromen-4-one Derivatives Against Chikungunya Virus. Lett. Drug Des. Discov. 2016, 13, 1019–1024.

