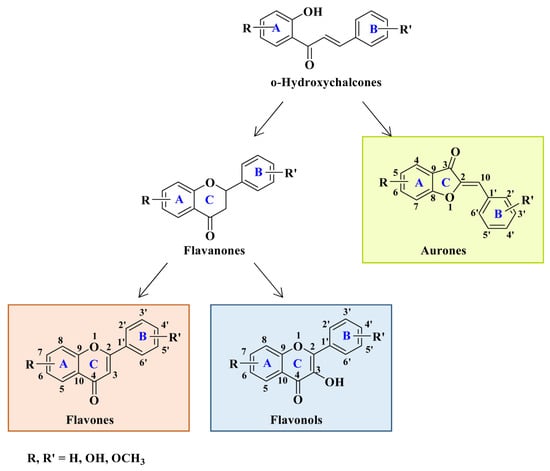
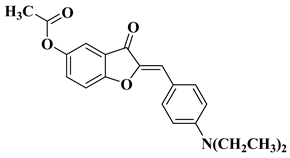
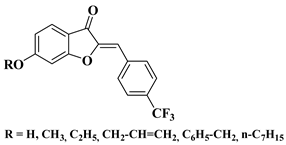
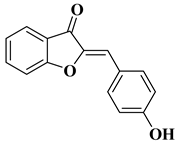
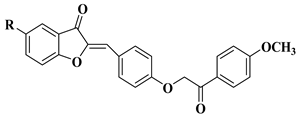
The outstanding biological potential of natural flavonoids has attracted interest in the medical field, meaning that many of their synthetic analogs are currently known as promising candidates in treatments for cancer; microbial, fungal, and viral infections; inflammatory diseases; and diabetes.
2. Anticancer Activity
The antitumor activity of flavones is most often due to their ability to target certain key structures that lead to cell cycle arrest and the apoptosis of tumor cells. Thus, flavones can inhibit the specific enzymes responsible for tumorigenesis, which are normally involved in the regulation of the cell cycle but whose function is deregulated under pathological conditions, for example, protein kinase C (PKC) [
5], cyclin-dependent kinases (CDK) [
6], casein kinases (CK) [
7], PIM-1 kinases [
8], death-associated protein kinase 1 (DAPK-1), and tyrosine kinases [
9]. Some flavones can inhibit the polymerization of tubulin, thus preventing the formation of microtubules [
10]. All this leads to cell cycle arrest, most often in the G2/M phase. Flavones can also activate certain enzymes that cause tumor cell apoptosis, such as caspases [
11,
12].
Natural flavones such as apigenin and nobiletin can regulate the expression of important inflammatory signaling pathways, including nuclear factor erythroid 2-related factor 2 (Nrf2) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The antioxidant properties of several natural flavones are attributable to their ability to regulate the expression of Nrf2/heme oxygenase-1 (HO-1), which decreases free radical levels and oxidative stress [
13]. Nuclear factor erythroid 2-related factor (Nrf2) can interact with the NF-κB signaling pathway to maintain cellular redox homeostasis during inflammatory states. As the NF-κB pathway activates the expression of genes implicated in inflammation that can lead to chronic inflammation, tumor development, or proliferation, the Nrf2 pathway displays important antioxidant roles, such as mediating the release of ROS (reactive oxygen species) induced by NF-κB or suppressing the transcription of NF-κB-dependent pro-inflammatory genes [
14]. Thus, the activation of Nrf2 pathway will suppress the NF-κB pathway and reduce TNFα, IL-6, and IL-1β proinflammatory cytokine levels [
14].
The potential of flavones to act on multiple anticancer targets or by synergic mechanisms of action allows them to be considered as key structures for the development of new multitarget-acting therapeutic agents.
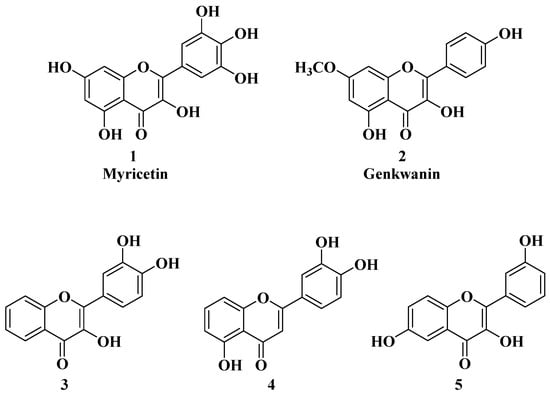
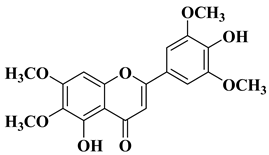
In several cases, the anticancer activity of natural flavones and aurones is closely related to their antioxidant activity. Myricetin (
Figure 2), a natural flavone with polyphenol structure, presents good antioxidant properties by acting as a scavenger for reactive oxygen species and by enhancing the activity of glutathione-
S-transferase [
15]. Myricetin also presents great antitumor properties by targeting key structures, leading to cell cycle arrest and apoptosis. Myricetin has been shown to inhibit several enzymes involved in cell cycle regulation whose functions were deregulated under pathological conditions, namely, PKC, CK2, PIM-1, and DAPK1 [
16]. Myricetin promotes tumor cell apoptosis by modulating certain signaling pathways, including Bcl2 (B-cell lymphoma 2), NF-κB, MAPKs (mitogen-activated protein kinases), and the Wnt/β-catenin signaling pathway [
16,
17,
18]. Recently, it was reported that myricetin inhibits interferon-γ-induced programmed death ligand-1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO1) expression in lung cancer cells via the regulation of the Janus kinase/signal transducer and activator of the JAK/STAT-IRF1 transcription pathway [
19]. According to the authors of [
19], in their study, Myricetin recovered the function of T cells in the lung cancer cells and Jurkat-PD-1 T cells. Myricetin restored the survival, proliferation, CD69 expression, and interleukin-2 (IL-2) secretion of Jurkat-PD-1 T cells suppressed by IFN-γ-treated lung cancer cells [
19]. PD-L1 and ISO1 are two immune checkpoints responsible for the immune escape of tumors. Thus, as an inhibitor of IFN-γ-induced PD-L1 and ISO1, myricetin has potential applications in tumor immunotherapy.
Recent studies have shown that myricetin induces apoptosis and autophagy in human gastric cancer cells through the inhibition of the PI3K/Akt/mTOR pathway (phosphoinositide 3-kinase, PI3K/Protein kinase B, Akt/Mechanistic target of rapamycin, mTOR) [
20]. The abnormal increase in the activity of the PI3K/Akt/mTOR pathway is associated with various malignancies; therefore, the modulation of this signaling pathway represents a new strategy, in particular in gastric cancer treatment [
21].
Myricetin also proved to be effective in preventing mutagenesis induced by different carcinogenic compounds such as formaldehyde [
22]. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through the inhibition of human hypoxia-inducible factor 1 subunit alpha (HIF-1α), an important target in lung and ovarian tumors [
22].
Gu Ling et al. recently revealed that myricetin regulates the p38 MAPK pathway by targeting MAP Kinase Kinase 3 (MKK3) in non-small cell lung cancer cells (NSCLC) [
23]. These results encourage future research on the development of new anticancer agents, MKK3 inhibitors, through the structural modulation of myricetin.
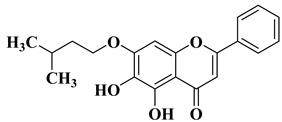
Genkwanin (
Figure 2), another natural flavone with antioxidant properties, has demonstrated promising anticancer activity against a series of cancer cell lines, including human MCF-7 breast cancer (IC
50 = 13.6 ± 0.3 µg/mL), HepG-2 human hepatocellular carcinoma (IC
50 = 22.5 ± 0.3 µg/mL), and HCT-116 colon cancer (IC
50 = 15.4 ± 0.5 µg/mL). Genkwanin is also able to reduce the migration, invasion, and proliferation of lung cancer cells by targeting the phosphoinositide 3-kinase (PI3K) and phospho-protein kinase B (AKT) signaling pathways [
24]. Due to this mechanism, genkwanin represents an effective option for the treatment of cancer proliferation and metastasis. Because genkwanin presents low oral bioavailability, genkwanin nanosuspensions were prepared in order to improve its solubility and pharmacokinetic profile. Li Y et al. reported the therapeutic potential of genkwanin nanosuspensions as novel antitumor agents in breast carcinoma therapy [
25].
Spiegel M. et al. established that, through the bond dissociation enthalpy (BDE) of the hydrogen atom transfer (HAT) mechanism, the antioxidant activity of flavones could be related to the presence of a hydroxy group located on the B ring, especially in position C4′, more than the A-ring substitution. Regarding flavonols, the presence of a hydroxy group in C3 is beneficial for their antioxidant activity. These positions present the lowest values of bond dissociation entalphy (BDE = 84.4 kcal/mol for C4′, in the case of luteolin, and BDE = 84.6 kcal/mol for C3, in the case of morin) [
26].
The anticancer activity of flavones could be correlated with their antioxidant activity, but it is not a mandatory rule in all cases. Grigalius I. and Petrikaite V. studied the relationship between the anticancer and antioxidant activities of trihydroxyflavones. The antioxidant activity was evaluated by using the DPPH (2,2-diphenyl-1-picrylhydrazyl) method, and the anticancer activity was evaluated by using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) method, both of which were performed on three different types of human cancer cell lines: lung (A549), breast (MCF-7), and brain epithelium (U87). Based on the calculation of the Pearson coefficient (r), a moderate correlation was revealed between the two biological properties [
27]. It was found that the substituents on the phenyl ring (B ring) are the most important for the antioxidant activity of trihydroxyflavones. Thus, the most potent antioxidants have the
o-dihydroxy group (catechol) on the B ring and are involved in binding hydroxy, peroxyl, and peroxynitrile radicals [
27] (
Figure 2, compounds
3 and
4). However, hydroxyflavone
5 does not possess this structural feature, but it does present the best anticancer activity, thus, in this case, alluding to the existence of other mechanisms of action for anticancer activity besides the neutralizing effect of free radicals.
Figure 2. Polyphenolic flavones with anticancer and antioxidant activity [
24,
27].
Zhao L. et al studied the structural elements of flavones capable of blocking different serine-threonine kinases involved in the cell cycle. Structure–activity relationship studies were conducted for PKC, CK-2, PIM-1 kinase, DAPK-1, and CDK. It was found that the hydroxy groups grafted on rings A, B, and C act as H-bond donors/acceptors in the interaction with PKC, PIM-1, DAPK-1, and CDK [
28]. For the inhibition of CK-2, it was found that the presence of the halogen atoms Br and Cl at positions 6 and 8 of the A ring, respectively, and the hydroxy group only in position 4 of the B ring are beneficial. The carbonyl group located in position 4 of the chromen-4-one ring acts as a H-bond acceptor in the interaction with various amino acid residues from CK-2, CDK, and PIM-1. The benzene ring (B ring) interacts by π–π stacking with the phenylalanine residue Fen113 of CK-2, and through this mechanism, it also blocks the ATP binding site of these enzymes. The benzene ring (B ring) can also make van der Waals interactions with certain hydrophobic residues from CDK-9 and PIM-1, thus making additional contact with these enzymes without blocking the binding of ATP. It has also been observed that changing the position of the phenyl ring from 2 to 3, specific to isoflavones, leads to the loss of activity [
28].
Flavopiridol (
Table 1, line 1) is a semisynthetic flavone that is currently being used in clinical trials as an anticancer agent for the treatment of acute myeloid leukemia. This compound acts by inhibiting kinases CDK-1, -2, -4, -6, and -7, all of which are competitive with ATP. At the same time, flavopiridol significantly inhibits kinases CDK-9 (non-competitive with ATP) [
9,
28]. Flavopiridol also inhibits the activity of positive transcription elongation factor (P-TEFb), a cyclin-dependent kinase controlling elongation via RNA polymerase II [
29].
The anticancer activity of flavopiridol is due to the presence of a chromone moiety that is bioisosteric with the purine ring of ATP and binds competitively to the ATP binding pocket of CDK. The benzene ring (ring B) provides additional contact with the enzyme, as it interacts with different regions than those occupied by ATP, participating in van der Waals-type interactions with other amino acid units [
28]. Other important elements for the inhibition of kinase activity by flavopiridol are the hydroxy groups at C-7 and C-5, the carbonyl group at C-4, the nitrogen atom, and the hydroxy group from the piperidine, and all of these functional groups are involved in the formation of hydrogen bonds with CDK [
28].
Starting from the structures of two natural products with anticancer activity with different mechanisms of action, 3,5,4′-trimethoxystilbene and 5,6,7-trimethoxyflavone, Hassan A.H. et al. synthesized new antiproliferative compounds by combining two pharmacophore moieties in the same molecule by replacing the vinylene residue in stilbene with the amide group [
30]. The cytotoxic activities of the synthesized compounds against several cancer cell lines were determined at 10 μM doses in all cases. The structures of the most active compounds are presented in
Table 1, lines 2–4.
Flavone–stylbene hybrids in which the nitrogen atom of the amide linker is attached to the flavone moiety proved to be more citotoxic than the corresponding compounds with the opposite amide linker configuration. Trimethoxylated flavone–stylbene hybrids showed superior activity compared to dimethoxylated flavone–stylbene hybrids on hematologic, colorectal, central nervous system, ovarian, renal, and breast cancer cell lines [
30]. On lung cancer cell lines, the dimethoxylated derivatives were generally more active than the trimethoxylated ones. Most of the tested hybrid compounds showed selective activity, showing no cytotoxicity on normal cells. Their anticancer mechanism of action consists of inducing apoptosis and inhibiting cell proliferation [
30].
Continuing their research, Hassan A.H. et al. synthesized a series of trimethoxyflavone-based aryl-amides, starting from the structures of already approved arylamide-type medicinal compounds (imatinib, masitinib) and replacing the bulky aromatic entity in their structure with 5,6,7-trimethoxyflavone and 5-hydroxy-6,7-dimethoxyflavone. The formation of the amide bond was carried out in the 3′ and 4′ positions on the B ring of the flavone using 3′-amino and 4′-amino precursors coupled with various acyl chlorides and 3′-carboxyl precursors condensed with aryl amines, respectively [
31].
Two flavones presented good broad-spectrum anticancer activity by triggering cell cycle arrest in the G1 phase (
Table 1, line 5). These compounds could represent hit compounds for the design of new, more potent inhibitors of STE20/GCK-IV kinase family members, including HGK, TNIK, and MINK1 kinases. It was found that the presence of the carbonyl of the amide linker attached to the flavone moiety is beneficial for the anticancer activity of the tested flavone-based aryl-amides. Reversing the attachment mode of the amide linker led to a significant decrease in anticancer activity [
31].
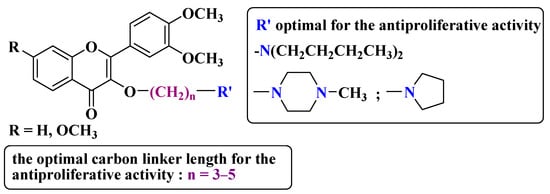
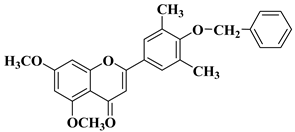
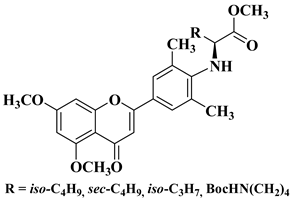
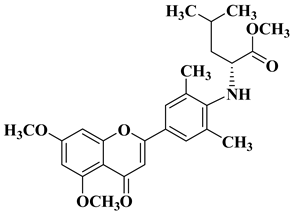
A series of dimethoxyflavonols and trimethoxyflavonols derivatives were obtained via the alkylation of the hydroxy group at position 3 of the chromen-4-one ring (C ring) (
Figure 3). The compounds were investigated for their anticancer activity on both androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU145) prostate cancer cell lines [
32].
Figure 3. 3-
O-substituted flavonols reported as anti-prostate cancer agents. Structure–activity relationship study [
32].
It was found that the alkylation of the hydroxy group in position 3 generally increased the antiproliferative activity of the compounds. The presence of an amino group linked to the hydroxy group at position 3 of the flavonols through a three- to five-carbon linker is beneficial for antiproliferative activity against the three human prostate cancer cell lines with tumor selectivity.
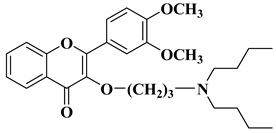
N-methylpiperazin-1-yl, pyrrolidin-1-yl, and dibutyl amino groups proved to be beneficial, improving the anticancer activity of the tested compounds. The most promising derivative in terms of selectivity, anticancer activity, and bioavailability contains a dibutyl amino group linked to the oxygen at position 3 via a three-carbon linker (
Table 1, line 6). The bioavailability of the tested compounds was superior to that of fisetin [
32].
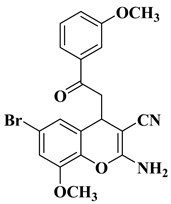
Starting from a series of differently substituted chalcones, Pontes et al. synthesized a series of chromene–chalcone hybrid compounds in order to test their anticancer activity on breast cancer cell lines. The most active compound is depicted in
Table 1, line 7. The mechanism of action involves the inhibition of cell migration and induction of apoptosis, by determining cell cycle arrest in the G2/M phase. Moreover, this compound has been proved to alter tubulin polymerization, representing a promising new microtubule-destabilizing agent. It was found that the presence of the halogen atoms grafted on the basic skeleton of chromene–chalcone hybrids is beneficial to antitumor activity. Brominated compounds presented superior activity to chlorinated and fluorinated compounds. The evaluated compounds presented selective cytotoxicity on cancer cell lines compared to non-cancerous cell lines [
10].
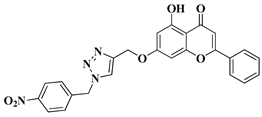
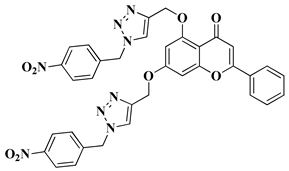
New hybrid compounds of flavones (chrysin and kaempferol) and substituted 1,2,3-triazoles were recently synthesized via the chemical derivatization of the hydroxyl groups of chrysin and kaempferol with functionalized 1,2,3-triazole compounds [
33]. The antitumor activity of the obtained mono- and bis-coupled hybrids was evaluated in vitro on 60 cell lines of 9 common cancer types (NCI60) [
33]. The hybrid compounds presenting the most significant antiproliferative effect are mentioned in
Table 1, lines 8, 9.
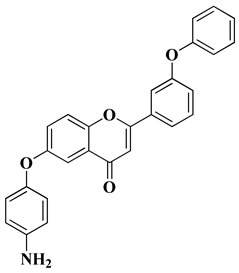
A series of new heterocyclic derivatives were recently synthesized via the functionalization of a flavone ring with an aminophenoxy moiety in different positions of the A ring and a phenoxy moiety in different positions of the B ring [
34]. Their cytotoxicity was investigated in vitro against two human non-small cell lung cancer (NSCLC) cell lines (A549 and NCI-H1975). It was found that the presence of a 4-aminophenoxy group at the sixth position of the A ring and a terminal phenoxy group on the B ring is beneficial for cancer-selective cytotoxicity. A flavone derivative containing a phenoxy moiety at the C’3 position of the B ring and a
p-aminophenoxy group at the sixth position of the A ring was the most effective, presenting micromolar IC
50 values (for A549 and H1975) and a high selectivity index (SI > 10,
Table 1, line 10). Further flow cytometric analyses showed that this compound induces apoptosis and cell cycle arrest in the G2/M phase through the up-regulation of p21 expression [
34]. The absence of the phenoxy moiety on the B ring and the different position of the
p-aminophenoxy moieties on the A ring decreased the efficacy and selectivity of aminophenoxy derivatives [
34].
New
C-dimethylated flavones were recently synthesized and evaluated for their anti-tubercular and anticancer activity [
35]. In this study, four flavones presented anticancer activity against a human adenocarcinoma A549 cell line, with IC
50 values between 39 and 48 μM (
Table 1, lines 11–14). This study’s in silico docking simulations revealed that these four compounds present improved binding and interaction profiles against the epidermal growth factor receptor (EGFR) [
35].
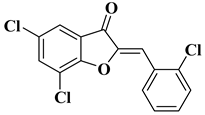
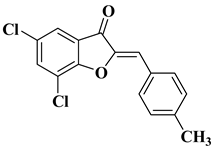
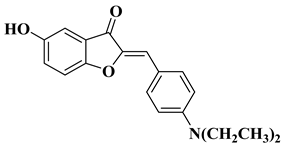
Other recently reported examples of synthetic flavones with antitumor activity are illustrated in Table 1, lines 15–20.
Natural and synthetic aurones possess a broad variety of biological activities, including antiproliferative activity against different cancer cell lines. The anticancer activity of aurones is due to their ability to interact with different key antitumor molecular targets, and examples of such interactions include the following: the inhibition of serine/threonine cyclin-dependent kinases (CDK 1 and 2) [
36], the inhibition of topoisomerase IIα [
37], the inhibition of sphingosine kinase (SphK) [
38], and interfering with microtubule assembly [
39]. In some cases, it was found that the anticancer activity of aurones is strongly related to their antioxidant activity [
40].
Several aurones have been shown to modulate the activity of ATP-dependent efflux pumps such as P-glycoprotein [
41] and breast cancer resistance protein (BCRP/ABCG2) [
42]. Through this mechanism, aurones can potentiate the effect of simultaneously administered anticancer chemotherapeutics by blocking the multidrug resistance mechanisms of tumor cells.
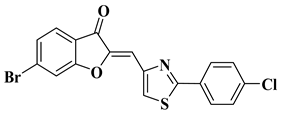
Our research group synthesized a series of aurone analogs by replacing the B ring (phenyl) with the 2-arylthiazole system in order to obtain compounds with superior anticancer activity, considering the anticancer potential of thiazole derivatives. Two aurone analogs were active against cancer cell lines resistant to currently used chemotherapeutics, such as multidrug-resistant leukemia cell lines and breast cancer cell lines, and both showed cytotoxic activities that were superior to doxorubicin (
Table 1, lines 21, 22) [
43]. Other recently reported examples of synthetic aurones with antitumor activity are illustrated in
Table 1, lines 23–31.
Table 1. Synthetic analogs of flavones and aurones with antitumor properties.
3. Antibacterial and Antifungal Activity
Bacterial and fungal resistance to existing antibiotics is a worldwide health issue, particularly affecting the immunocompromised patients. Without effective antimicrobial agents, several medical procedures could endanger the lives of patients by increasing the risk of microbial infections. The basic structure of natural flavones and aurones have inspired researchers to develop new antimicrobial agents with improved bioavailability and antibacterial and antifungal properties.
Recently, it was reported that the natural flavone myricetin (
Figure 2) presents anti biofilm activity against
Staphylococcus aureus and attenuates osteomyelitis by inhibiting the Toll-like receptor-2 (TLR2)/mitogen-activated protein kinase (MAPK) pathway in experimental mice [
54].
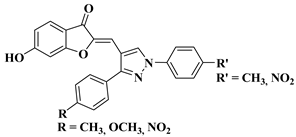
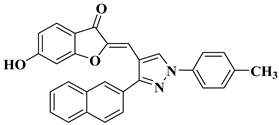
Ashok D. et al. synthesized new flavonol analogs bearing the extended heteroaromatic system 1-phenyl-3-(thiophen-2-yl)-1
H-pyrazol-4-yl instead of a phenyl ring (B ring) and containing various substituents on the chromone system. The synthesized flavonol derivatives were screened for their antimicrobial activity against several fungal strains (
Aspergillus niger,
Penicillium italicum,
Fusarium oxysporum) and bacterial strains (
Staphylococcus aureus,
Pseudomonas aeruginosa,
Escherichia coli,
Bacillus subtilis). The inhibition zones (IZ) were determined at 50 μg/mL concentration for each compound in dimethyl sulfoxide (used as a solvent). Four of the tested compounds (
Table 2, lines 1–4) show good antimicrobial activity and represent hit compounds for the design of new antifungal and/or antibacterial therapeutic agents [
55].
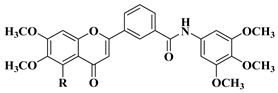
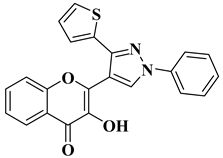
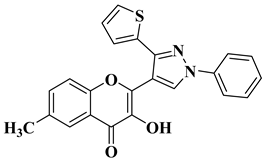
In order to obtain new flavone analogs with antibacterial activity, Lv X.H. et al. synthesized a series of flavone Mannich base derivatives by applying the Mannich reaction between primary amines and using natural flavones as components with mobile hydrogen and formaldehyde as a carbonyl component. The natural flavones used as precursors were baicalein, luteolin, quercetol, apigenin, and kaempferol. Derivatization was performed at position 8 of the chromone moiety by applying the Mannich reaction [
56]. The antibacterial activity of the obtained flavone Mannich bases was evaluated for two Gram-positive bacteria (
S. aureus and
Listeria monocytogenes) and two Gram-negative bacteria (
E. coli and
Salmonella gallinarium), and novobiocin and ciprofloxacin were used as standards. The structures of the most active compounds are shown in
Table 2, lines 5, 6. Through performing in vitro experiments and in silico molecular docking studies, it was found that these compounds exhibit potent inhibition against topoisomerase II and topoisomerase IV isolated from
E. coli [
56].
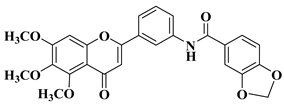
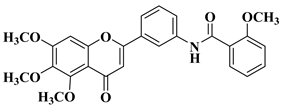
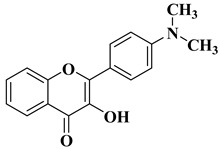
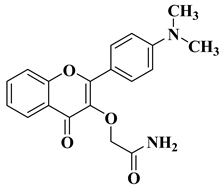
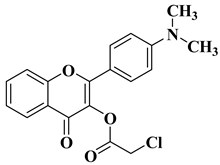
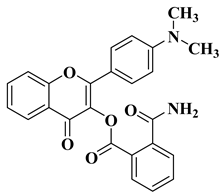
New hydroxyflavone derivatives containing the dimethylamino group grafted at position 4 of the benzene ring (B ring) were synthesized and evaluated for their antifungal activity against
Acremonium strictum,
Penicillium expansum, and
Aspergillus flavus. Four of the tested compounds presented very good antifungal activities against some of the tested fungal strains (
Table 2, lines 7–10) [
57].
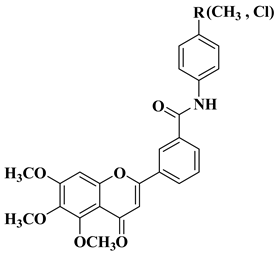
New quinoline-based aurone analogs were synthesized and evaluated for their antibacterial, antifungal, and anti-biofilm activity. The compounds mentioned in
Table 2, lines 11–13 presented the most significant antibacterial and antifungal activities, and some of them were also shown to be good anti-biofilm agents [
58].
New
C-dimethylated flavones were recently synthesized and evaluated for their anti-tubercular and anticancer activity [
35]. Two dimethylated and dimethoxylated flavones bearing the fluoro and dimethylamino substituents in position 4 of the B ring were shown to have significant antibacterial activity against the H37Rv strain of replicating
Mycobacterium tuberculosis, with sensitivity up to 6.25 µg/mL (
Table 2, line 14).
4. Antiviral Activity
Viral infections represent a global health issue and have had many implications on public health throughout history, including the appearance of new mutant viral strains and the emergence of pandemics. Specific aspects of modernization, such as rapid air transit and urbanization, have accelerated the emergence and spread of viruses. Antiviral therapy is necessary when vaccination does not bring the expected results or in the case of infections for which vaccination has not been implemented. Flavones have also been included in the research of new molecules with antiviral potential, yielding some important results and positive prospects for the future.
According to a recently reported study, the natural flavone myricetin (
Figure 2) possesses potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation through the RIPK1/NF-κB pathway [
59]. Myricetin also inhibits SASR-CoV-2 infection and replication in Vero E6 cells (EC
50 55.18 μM) [
59]; these results suggest that this flavone represents a key structure for the design of new therapeutic agents against COVID-19.
Regarding tricin, 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone, a flavone derivative with activity against cytomegalovirus (CMV), Fujimoto K.J. et al. modulated its structure by grafting a fluorine atom on the chromen-4-one ring. Thus, two compounds were obtained—6-F-tricin and 7-F-tricin—and the antiviral activity of which was measured against cytomegalovirus replicated on embryonic lung cell cultures. Compared to ganciclovir, 6-F-tricin showed much stronger activity against cytomegalovirus (
Table 2, line 15). Moreover, it was observed that 6-F-tricin did not produce cytotoxicity on the used embryonic cells. Substitution with fluorine is beneficial for increasing the affinity for target proteins (in this case, for CDK9, cyclin-dependent kinase 9) [
60].
The antiviral potential of flavones has also been demonstrated against tropical diseases such as Chikungunya fever. Badavath V.N. et al. synthesized nineteen flavones in order to evaluate their antiviral activity against Chikungunya virus replication. Two compounds showed activity at concentrations below 1 µg/mL (
Table 2, lines 16, 17). It was observed that the more potent compounds possess heterocycles (thiophen-2-yl and pyridyn-2-yl) in position 2 of the chromen-4-one ring instead of the benzene ring (B ring). Through conducting molecular docking studies, it was deduced that these compounds act by inhibiting the Chikungunya virus protease [
61].
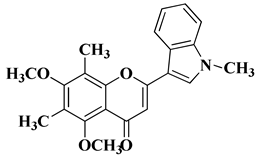
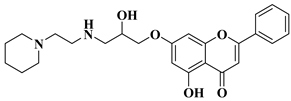
Table 2. Synthetic analogs of flavones and aurones with antimicrobial (antibacterial/antifungal/antiviral) properties.
|
Entry
|
Chemical Structure
|
Microbial Strains against the Tested Compounds Present Antimicrobial Activity
|
Ref.
|
|
1
|
|
Antifungal activity (inhibition zone for 50 μg/mL solution):
Aspergillus niger (IZ = 16 mm)
Penicillium italicum (IZ = 20 mm)
Fusarium oxysporum (IZ = 31 mm)
|
[55]
|
|
2
|
|
Antibacterial activity (inhibition zone for 50 μg/mL solution):
Staphylococcus aureus (IZ = 31 mm)
Pseudomonas aeruginosa (IZ = 11 mm)
Escherichia coli (IZ = 30 mm)
|
[55]
|
|
3
|
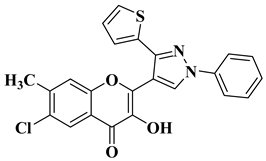
|
Antibacterial activity (inhibition zone for 50 μg/mL solution):
Staphylococcus aureus (IZ = 30 mm)
Bacillus subtilis (IZ = 11 mm)
Escherichia coli (IZ = 31 mm)
Antifungal activity (inhibition zone for 50 μg/mL solution):
Aspergillus niger (IZ = 13 mm)
Penicillium italicum (IZ = 24 mm)
Fusarium oxysporum (IZ = 25 mm)
|
[55]
|
|
4
|
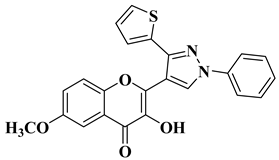
|
Antibacterial activity (inhibition zone for 50 μg/mL solution):
Staphylococcus aureus (IZ = 33 mm)
Bacillus subtilis (IZ = 17 mm)
Escherichia coli (IZ = 33 mm)
Antifungal activity (inhibition zone for 50 μg/mL solution):
Aspergillus niger (IZ = 14 mm)
Penicillium italicum (IZ = 26 mm)
Fusarium oxysporum (IZ = 27 mm)
|
[55]
|
|
5
|
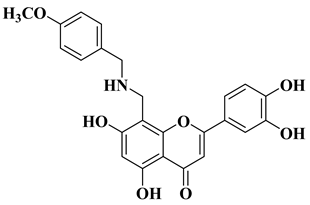
|
Antibacterial activity:
Staphylococcus aureus (MIC = 2 mg/L)
Escherichia coli (MIC = 4 mg/L)
Salmonella gallinarum (MIC = 0.125 mg/L)
|
[56]
|
|
6
|
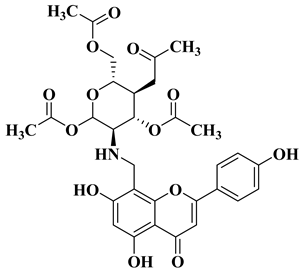
|
Antibacterial activity:
Staphylococcus aureus (MIC = 1 mg/L)
Escherichia coli (MIC = 2 mg/L)
Salmonella gallinarum (MIC = 0.05 mg/L)
Listeria monocytogenes (MIC = 0.5 mg/L)
|
[56]
|
|
7
|
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration):
Acremonium strictum (81.33%; 100%)
Penicillium expansum (60.87%; 100%)
Aspergillus flavus (41.02%; 65.64%)
|
[57]
|
|
8
|
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration):
Acremonium strictum (70%; 100%)
Penicillium expansum (42.15%; 100%)
Aspergillus flavus (6.41%; 46.15%)
|
[57]
|
|
9
|
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration):
Acremonium strictum (76.88%; 100%)
Aspergillus flavus (15.38%; 60.51%)
|
[57]
|
|
10
|
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration):
Acremonium strictum (73.33%; 100%)
|
[57]
|
|
11
|
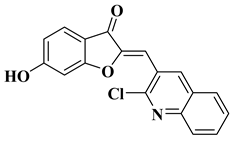
|
Antibacterial activity:
Staphylococcus aureus (MIC = 1.25 mg/mL)
Bacillus subtilis (MIC = 0.02 mg/mL)
Mycobacterium smegmatis (MIC = 0.625 mg/mL)
Antifungal activity:
Fusarium oxysporum (MIC = 0.625 mg/mL)
|
[58]
|
|
12
|
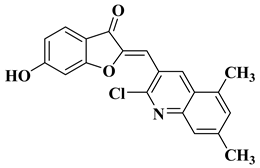
|
Antibacterial activity:
Staphylococcus aureus (MIC = 2.5 mg/mL)
Bacillus subtilis (MIC = 0.156 mg/mL)
Mycobacterium smegmatis (MIC = 0.078 mg/mL)
Anti biofilm and anti quorum sensing activity (100 μg/mL)
Antifungal activity:
Fusarium oxysporum (MIC = 0.313 mg/mL)
Candida albicans (MIC = 0.078 mg/mL)
|
[58]
|
|
13
|
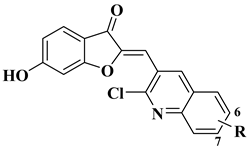
|
Antibacterial activity (R=6-OCH3, 7-Cl)
Staphylococcus aureus (MIC = 1.25 mg/mL)
Bacillus subtilis (MIC = 1.25 mg/mL)
Klebsiella pneumoniae (MIC = 0.625 mg/mL)
Anti biofilm activity (R=6-OCH3, 100 μg/mL)
Antifungal activity (R=7-Cl):
Candida albicans (MIC = 0.156 mg/mL)
|
[58]
|
|
14
|
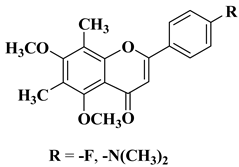
|
Antibacterial activity
Mycobacterium tuberculosis H37Rv
(MIC = 6.25 µg/mL)
|
[35]
|
|
15
|
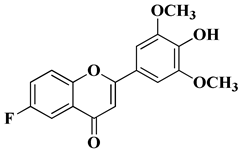
|
Antiviral activity
Human cytomegalovirus (EC50 = 0.126 nM)
|
[60]
|
|
16
|
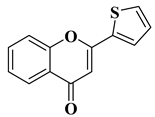
|
Antiviral activity
Chikungunya Virus (IC50 = 0.44 µM)
|
[61]
|
|
17
|
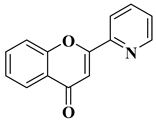
|
Antiviral activity
Chikungunya Virus (IC50 = 0.45 µM)
|
[61]
|