Flavonoids are a widely distributed group of natural polyphenolic compounds that are found in plants usually in glycosylated form and have been shown to possess a wide range of biological activities, including antioxidant, anti-inflammatory, antibacterial, antiviral, and anticancer properties, making them an attractive target for synthesis and further research.
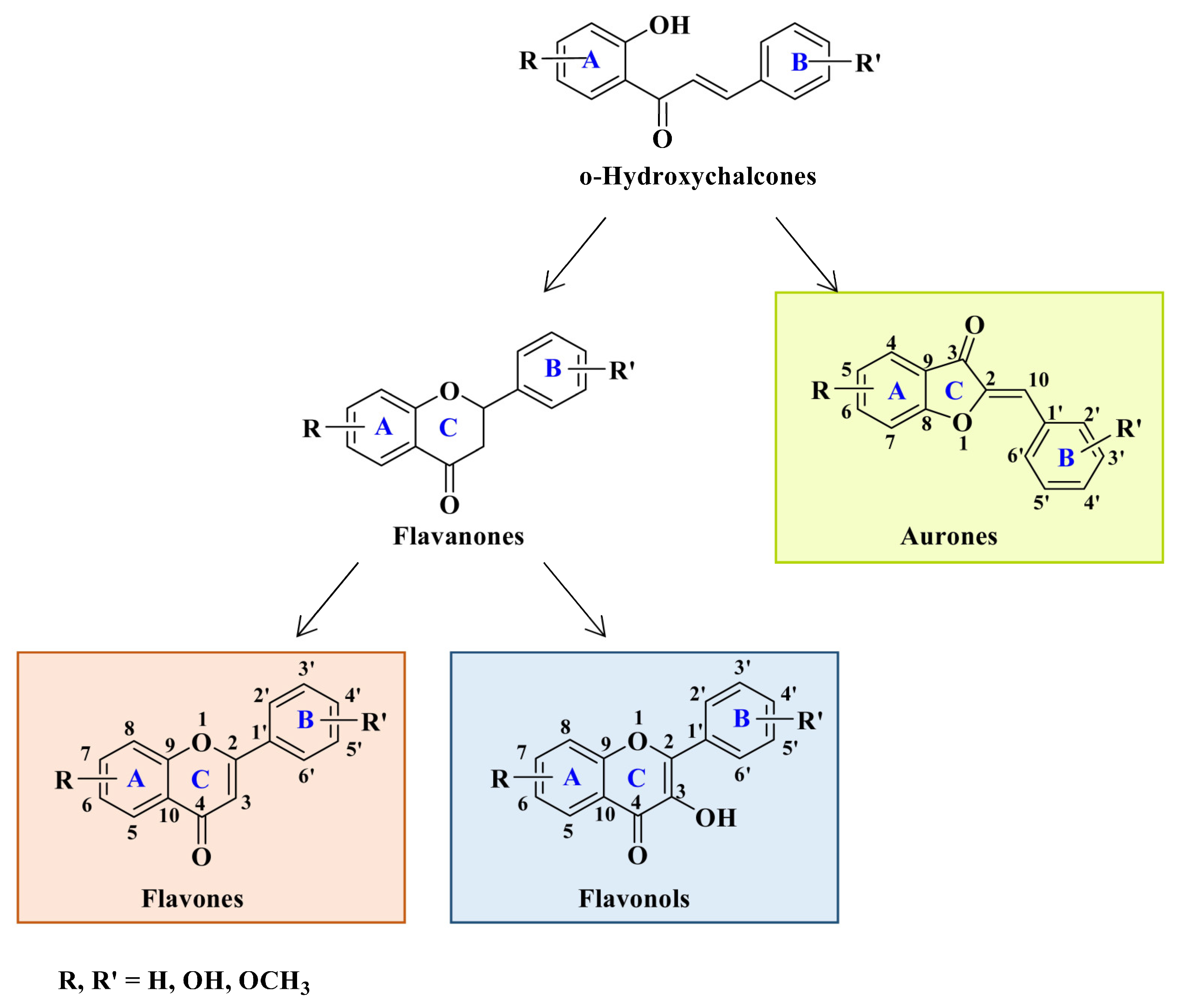
- chalcones
- flavones
- flavonols
- aurones
- anticancer activity
- antimicrobial activity
1. Introduction
2. Anticancer Activity
The antitumor activity of flavones is most often due to their ability to target certain key structures that lead to cell cycle arrest and the apoptosis of tumor cells. Thus, flavones can inhibit the specific enzymes responsible for tumorigenesis, which are normally involved in the regulation of the cell cycle but whose function is deregulated under pathological conditions, for example, protein kinase C (PKC) [5][3], cyclin-dependent kinases (CDK) [6][4], casein kinases (CK) [7][5], PIM-1 kinases [8][6], death-associated protein kinase 1 (DAPK-1), and tyrosine kinases [9][7]. Some flavones can inhibit the polymerization of tubulin, thus preventing the formation of microtubules [10][8]. All this leads to cell cycle arrest, most often in the G2/M phase. Flavones can also activate certain enzymes that cause tumor cell apoptosis, such as caspases [11,12][9][10]. Natural flavones such as apigenin and nobiletin can regulate the expression of important inflammatory signaling pathways, including nuclear factor erythroid 2-related factor 2 (Nrf2) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The antioxidant properties of several natural flavones are attributable to their ability to regulate the expression of Nrf2/heme oxygenase-1 (HO-1), which decreases free radical levels and oxidative stress [13][11]. Nuclear factor erythroid 2-related factor (Nrf2) can interact with the NF-κB signaling pathway to maintain cellular redox homeostasis during inflammatory states. As the NF-κB pathway activates the expression of genes implicated in inflammation that can lead to chronic inflammation, tumor development, or proliferation, the Nrf2 pathway displays important antioxidant roles, such as mediating the release of ROS (reactive oxygen species) induced by NF-κB or suppressing the transcription of NF-κB-dependent pro-inflammatory genes [14][12]. Thus, the activation of Nrf2 pathway will suppress the NF-κB pathway and reduce TNFα, IL-6, and IL-1β proinflammatory cytokine levels [14][12]. The potential of flavones to act on multiple anticancer targets or by synergic mechanisms of action allows them to be considered as key structures for the development of new multitarget-acting therapeutic agents. In several cases, the anticancer activity of natural flavones and aurones is closely related to their antioxidant activity. Myricetin (Figure 2), a natural flavone with polyphenol structure, presents good antioxidant properties by acting as a scavenger for reactive oxygen species and by enhancing the activity of glutathione-S-transferase [15][13]. Myricetin also presents great antitumor properties by targeting key structures, leading to cell cycle arrest and apoptosis. Myricetin has been shown to inhibit several enzymes involved in cell cycle regulation whose functions were deregulated under pathological conditions, namely, PKC, CK2, PIM-1, and DAPK1 [16][14]. Myricetin promotes tumor cell apoptosis by modulating certain signaling pathways, including Bcl2 (B-cell lymphoma 2), NF-κB, MAPKs (mitogen-activated protein kinases), and the Wnt/β-catenin signaling pathway [16,17,18][14][15][16]. Recently, it was reported that myricetin inhibits interferon-γ-induced programmed death ligand-1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO1) expression in lung cancer cells via the regulation of the Janus kinase/signal transducer and activator of the JAK/STAT-IRF1 transcription pathway [19][17]. According to the authors of [19][17], in their study, Myricetin recovered the function of T cells in the lung cancer cells and Jurkat-PD-1 T cells. Myricetin restored the survival, proliferation, CD69 expression, and interleukin-2 (IL-2) secretion of Jurkat-PD-1 T cells suppressed by IFN-γ-treated lung cancer cells [19][17]. PD-L1 and ISO1 are two immune checkpoints responsible for the immune escape of tumors. Thus, as an inhibitor of IFN-γ-induced PD-L1 and ISO1, myricetin has potential applications in tumor immunotherapy. Recent studies have shown that myricetin induces apoptosis and autophagy in human gastric cancer cells through the inhibition of the PI3K/Akt/mTOR pathway (phosphoinositide 3-kinase, PI3K/Protein kinase B, Akt/Mechanistic target of rapamycin, mTOR) [20][18]. The abnormal increase in the activity of the PI3K/Akt/mTOR pathway is associated with various malignancies; therefore, the modulation of this signaling pathway represents a new strategy, in particular in gastric cancer treatment [21][19]. Myricetin also proved to be effective in preventing mutagenesis induced by different carcinogenic compounds such as formaldehyde [22][20]. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through the inhibition of human hypoxia-inducible factor 1 subunit alpha (HIF-1α), an important target in lung and ovarian tumors [22][20]. Gu Ling et al. recently revealed that myricetin regulates the p38 MAPK pathway by targeting MAP Kinase Kinase 3 (MKK3) in non-small cell lung cancer cells (NSCLC) [23][21]. These results encourage future research on the development of new anticancer agents, MKK3 inhibitors, through the structural modulation of myricetin. Genkwanin (Figure 2), another natural flavone with antioxidant properties, has demonstrated promising anticancer activity against a series of cancer cell lines, including human MCF-7 breast cancer (IC50 = 13.6 ± 0.3 µg/mL), HepG-2 human hepatocellular carcinoma (IC50 = 22.5 ± 0.3 µg/mL), and HCT-116 colon cancer (IC50 = 15.4 ± 0.5 µg/mL). Genkwanin is also able to reduce the migration, invasion, and proliferation of lung cancer cells by targeting the phosphoinositide 3-kinase (PI3K) and phospho-protein kinase B (AKT) signaling pathways [24][22]. Due to this mechanism, genkwanin represents an effective option for the treatment of cancer proliferation and metastasis. Because genkwanin presents low oral bioavailability, genkwanin nanosuspensions were prepared in order to improve its solubility and pharmacokinetic profile. Li Y et al. reported the therapeutic potential of genkwanin nanosuspensions as novel antitumor agents in breast carcinoma therapy [25][23]. Spiegel M. et al. established that, through the bond dissociation enthalpy (BDE) of the hydrogen atom transfer (HAT) mechanism, the antioxidant activity of flavones could be related to the presence of a hydroxy group located on the B ring, especially in position C4′, more than the A-ring substitution. Regarding flavonols, the presence of a hydroxy group in C3 is beneficial for their antioxidant activity. These positions present the lowest values of bond dissociation entalphy (BDE = 84.4 kcal/mol for C4′, in the case of luteolin, and BDE = 84.6 kcal/mol for C3, in the case of morin) [26][24]. The anticancer activity of flavones could be correlated with their antioxidant activity, but it is not a mandatory rule in all cases. Grigalius I. and Petrikaite V. studied the relationship between the anticancer and antioxidant activities of trihydroxyflavones. The antioxidant activity was evaluated by using the DPPH (2,2-diphenyl-1-picrylhydrazyl) method, and the anticancer activity was evaluated by using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) method, both of which were performed on three different types of human cancer cell lines: lung (A549), breast (MCF-7), and brain epithelium (U87). Based on the calculation of the Pearson coefficient (r), a moderate correlation was revealed between the two biological properties [27][25]. It was found that the substituents on the phenyl ring (B ring) are the most important for the antioxidant activity of trihydroxyflavones. Thus, the most potent antioxidants have the o-dihydroxy group (catechol) on the B ring and are involved in binding hydroxy, peroxyl, and peroxynitrile radicals [27][25] (Figure 2, compounds 3 and 4). However, hydroxyflavone 5 does not possess this structural feature, but it does present the best anticancer activity, thus, in this case, alluding to the existence of other mechanisms of action for anticancer activity besides the neutralizing effect of free radicals.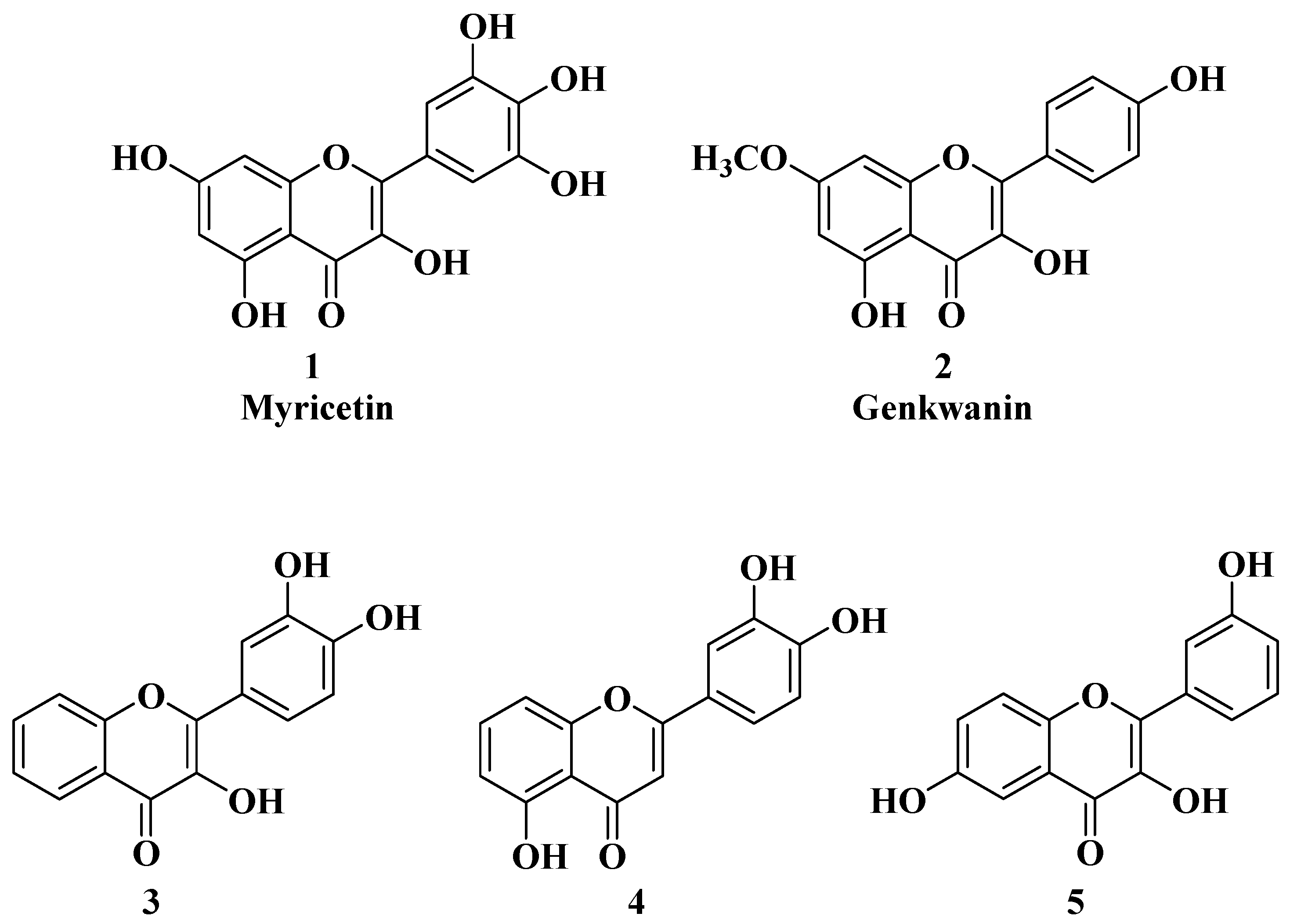
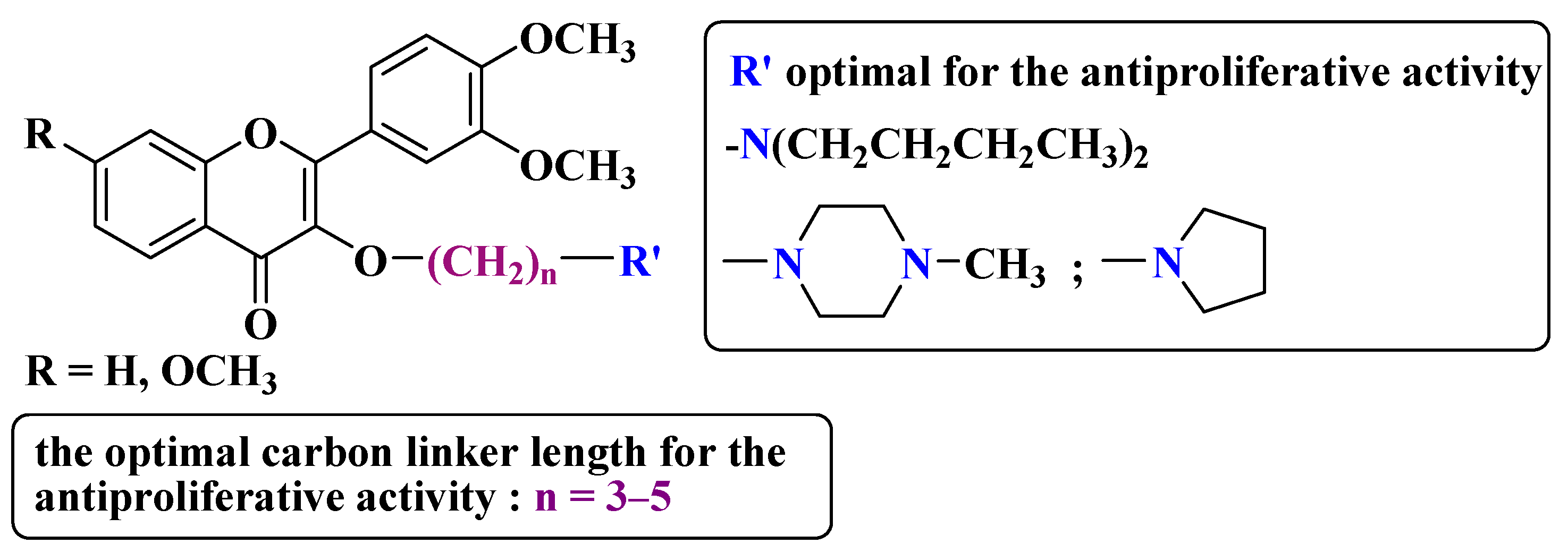
|
Entry |
Chemical Structure |
Cancer Cell Lines against the Tested Compounds Present Cytotoxic Activity |
Ref. |
|||
|---|---|---|---|---|---|---|
|
1 |
|
|
||||
|
2 |
|
R = OCH3
|
||||
|
R = OH
|
||||||
|
3 |
|
|
||||
|
4 |
|
|
||||
|
5 |
|
melanoma (160.26–107.81% SKMEL5), hematologic (111.12–92.74% leukemia HL60), renal (129.05% RXF393), colon (98.27–82.03% COLO205), lung (93.28% H522), brain (147.04–141.63% SF295 glioma), ovarian (76.54–51.79% IGROV1, OVCAR3, OVCAR8, ADREES, SKOV3), growth inhibition determined at 10 µM dosage. |
||||
|
6 |
|
|
||||
|
7 |
|
|
||||
|
8 |
|
|
||||
|
9 |
10
|
|
CNS cancer: SF-268 (GI50 = 3.52 µM), SF-295 (GI50 = 2.32 µM), SF-539 (GI50 = 2.21 µM), SNB-19 (GI50 = 4.55 µM), SNB-75 (GI50 = 1.69 µM), U251 (GI50 = 2.80 µM).
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (73.33%; 100%)
|
||
[ | ][55] |
10 |
||||
|
11 |
|
|
|
|||
Antibacterial activity: |
Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 0.02 mg/mL) Mycobacterium smegmatis (MIC = 0.625 mg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.625 mg/mL) |
11 |
||||
|
12 |
|
|
|
Antibacterial activity: Staphylococcus aureus (MIC = 2.5 mg/mL) Bacillus subtilis (MIC = 0.156 mg/mL) Mycobacterium smegmatis (MIC = 0.078 mg/mL) Anti biofilm and anti quorum sensing activity (100 μg/mL) Antifungal activity: Fusarium oxysporum (MIC = 0.313 mg/mL) | ||
Candida albicans | (MIC = 0.078 mg/mL) |
12 |
||||
|
13 |
|
|
|
|
||
|
13 |
|
|
||||
|
14 |
|
|
||||
|
15 |
|
|
||||
|
16 |
|
|
||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
| ||||||
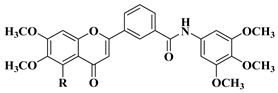
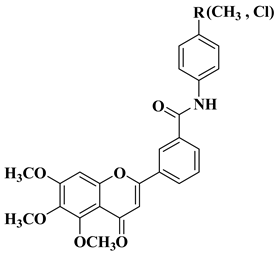
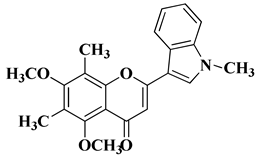
|
29 |
|
R = Cl: leukemia cell lines MOLT-4 (−17.79% mean growth percentage), and SR (−22.38% mean growth percentage). R = H: renal cancer cell line UO-31 (−44.36% mean growth percentage). The mean growth percentages were determined for five concentrations ranging from 10−4 to 10−8 M. |
||||
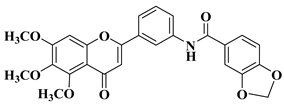
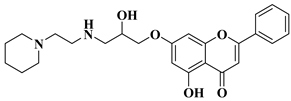
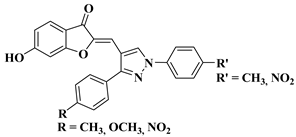
|
30 |
|
R′ = CH3 and R = CH3, OCH3, NO2 (IC50 = 25–28.3 µM). R′ = NO2 and R = NO2 (IC50 = 25.1 µM). |
||||
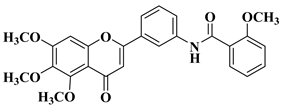
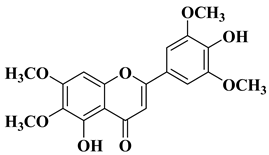
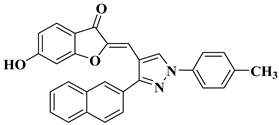
|
31 |
|
|
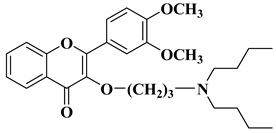
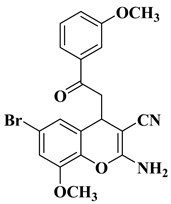
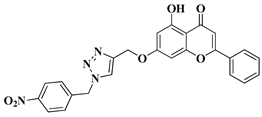
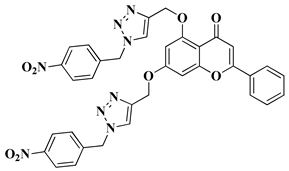
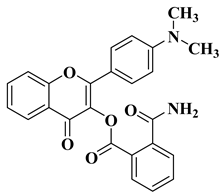
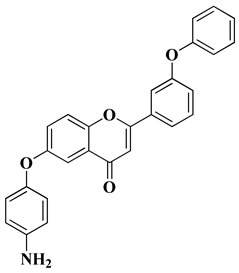
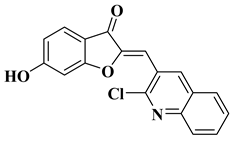
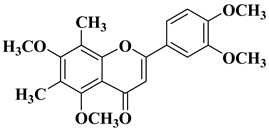
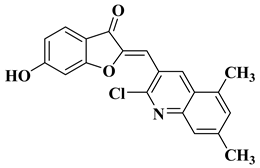
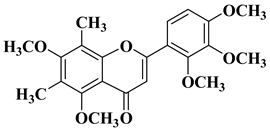
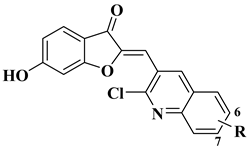
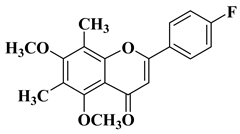
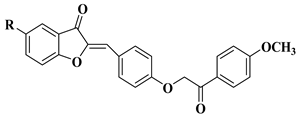
3. Antibacterial and Antifungal Activity
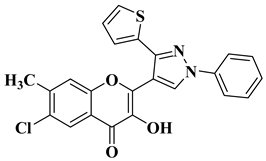
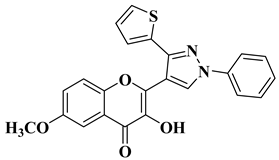
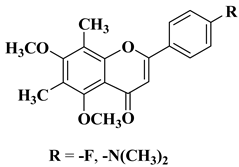
Bacterial and fungal resistance to existing antibiotics is a worldwide health issue, particularly affecting the immunocompromised patients. Without effective antimicrobial agents, several medical procedures could endanger the lives of patients by increasing the risk of microbial infections. The basic structure of natural flavones and aurones have inspired researchers to develop new antimicrobial agents with improved bioavailability and antibacterial and antifungal properties. Recently, it was reported that the natural flavone myricetin (Figure 2) presents anti biofilm activity against Staphylococcus aureus and attenuates osteomyelitis by inhibiting the Toll-like receptor-2 (TLR2)/mitogen-activated protein kinase (MAPK) pathway in experimental mice [54][52]. Ashok D. et al. synthesized new flavonol analogs bearing the extended heteroaromatic system 1-phenyl-3-(thiophen-2-yl)-1H-pyrazol-4-yl instead of a phenyl ring (B ring) and containing various substituents on the chromone system. The synthesized flavonol derivatives were screened for their antimicrobial activity against several fungal strains (Aspergillus niger, Penicillium italicum, Fusarium oxysporum) and bacterial strains (Staphylococcus aureus, Pseudomonas aeruginosa, Escherichia coli, Bacillus subtilis). The inhibition zones (IZ) were determined at 50 μg/mL concentration for each compound in dimethyl sulfoxide (used as a solvent). Four of the tested compounds (Table 2, lines 1–4) show good antimicrobial activity and represent hit compounds for the design of new antifungal and/or antibacterial therapeutic agents [55][53]. In order to obtain new flavone analogs with antibacterial activity, Lv X.H. et al. synthesized a series of flavone Mannich base derivatives by applying the Mannich reaction between primary amines and using natural flavones as components with mobile hydrogen and formaldehyde as a carbonyl component. The natural flavones used as precursors were baicalein, luteolin, quercetol, apigenin, and kaempferol. Derivatization was performed at position 8 of the chromone moiety by applying the Mannich reaction [56][54]. The antibacterial activity of the obtained flavone Mannich bases was evaluated for two Gram-positive bacteria (S. aureus and Listeria monocytogenes) and two Gram-negative bacteria (E. coli and Salmonella gallinarium), and novobiocin and ciprofloxacin were used as standards. The structures of the most active compounds are shown in Table 2, lines 5, 6. Through performing in vitro experiments and in silico molecular docking studies, it was found that these compounds exhibit potent inhibition against topoisomerase II and topoisomerase IV isolated from E. coli [56][54]. New hydroxyflavone derivatives containing the dimethylamino group grafted at position 4 of the benzene ring (B ring) were synthesized and evaluated for their antifungal activity against Acremonium strictum, Penicillium expansum, and Aspergillus flavus. Four of the tested compounds presented very good antifungal activities against some of the tested fungal strains (Table 2, lines 7–10) [57][55]. New quinoline-based aurone analogs were synthesized and evaluated for their antibacterial, antifungal, and anti-biofilm activity. The compounds mentioned in Table 2, lines 11–13 presented the most significant antibacterial and antifungal activities, and some of them were also shown to be good anti-biofilm agents [58][56]. New C-dimethylated flavones were recently synthesized and evaluated for their anti-tubercular and anticancer activity [35][33]. Two dimethylated and dimethoxylated flavones bearing the fluoro and dimethylamino substituents in position 4 of the B ring were shown to have significant antibacterial activity against the H37Rv strain of replicating Mycobacterium tuberculosis, with sensitivity up to 6.25 µg/mL (Table 2, line 14).4. Antiviral Activity
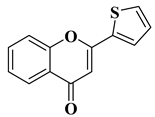
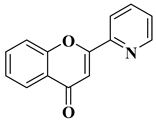
Viral infections represent a global health issue and have had many implications on public health throughout history, including the appearance of new mutant viral strains and the emergence of pandemics. Specific aspects of modernization, such as rapid air transit and urbanization, have accelerated the emergence and spread of viruses. Antiviral therapy is necessary when vaccination does not bring the expected results or in the case of infections for which vaccination has not been implemented. Flavones have also been included in the research of new molecules with antiviral potential, yielding some important results and positive prospects for the future. According to a recently reported study, the natural flavone myricetin (Figure 2) possesses potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation through the RIPK1/NF-κB pathway [59][57]. Myricetin also inhibits SASR-CoV-2 infection and replication in Vero E6 cells (EC50 55.18 μM) [59][57]; these results suggest that this flavone represents a key structure for the design of new therapeutic agents against COVID-19. Regarding tricin, 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone, a flavone derivative with activity against cytomegalovirus (CMV), Fujimoto K.J. et al. modulated its structure by grafting a fluorine atom on the chromen-4-one ring. Thus, two compounds were obtained—6-F-tricin and 7-F-tricin—and the antiviral activity of which was measured against cytomegalovirus replicated on embryonic lung cell cultures. Compared to ganciclovir, 6-F-tricin showed much stronger activity against cytomegalovirus (Table 2, line 15). Moreover, it was observed that 6-F-tricin did not produce cytotoxicity on the used embryonic cells. Substitution with fluorine is beneficial for increasing the affinity for target proteins (in this case, for CDK9, cyclin-dependent kinase 9) [60][58]. The antiviral potential of flavones has also been demonstrated against tropical diseases such as Chikungunya fever. Badavath V.N. et al. synthesized nineteen flavones in order to evaluate their antiviral activity against Chikungunya virus replication. Two compounds showed activity at concentrations below 1 µg/mL (Table 2, lines 16, 17). It was observed that the more potent compounds possess heterocycles (thiophen-2-yl and pyridyn-2-yl) in position 2 of the chromen-4-one ring instead of the benzene ring (B ring). Through conducting molecular docking studies, it was deduced that these compounds act by inhibiting the Chikungunya virus protease [61][59].|
Entry |
Chemical Structure |
Microbial Strains against the Tested Compounds Present Antimicrobial Activity |
Ref. |
||||
|---|---|---|---|---|---|---|---|
|
1 |
|
Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 16 mm) Penicillium italicum (IZ = 20 mm) Fusarium oxysporum (IZ = 31 mm) |
|||||
|
2 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 31 mm) Pseudomonas aeruginosa (IZ = 11 mm) Escherichia coli (IZ = 30 mm) |
|||||
|
3 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 30 mm) Bacillus subtilis (IZ = 11 mm) Escherichia coli (IZ = 31 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 13 mm) Penicillium italicum (IZ = 24 mm) Fusarium oxysporum (IZ = 25 mm) |
|||||
|
4 |
|
Antibacterial activity (inhibition zone for 50 μg/mL solution): Staphylococcus aureus (IZ = 33 mm) Bacillus subtilis (IZ = 17 mm) Escherichia coli (IZ = 33 mm) Antifungal activity (inhibition zone for 50 μg/mL solution): Aspergillus niger (IZ = 14 mm) Penicillium italicum (IZ = 26 mm) Fusarium oxysporum (IZ = 27 mm) |
|||||
|
5 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 2 mg/L) Escherichia coli (MIC = 4 mg/L) Salmonella gallinarum (MIC = 0.125 mg/L) |
|||||
|
6 |
|
Antibacterial activity: Staphylococcus aureus (MIC = 1 mg/L) Escherichia coli (MIC = 2 mg/L) Salmonella gallinarum (MIC = 0.05 mg/L) Listeria monocytogenes (MIC = 0.5 mg/L) |
|||||
|
7 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (81.33%; 100%) Penicillium expansum (60.87%; 100%) Aspergillus flavus (41.02%; 65.64%) |
|||||
|
8 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (70%; 100%) Penicillium expansum (42.15%; 100%) Aspergillus flavus (6.41%; 46.15%) |
|||||
|
9 |
|
Antifungal activity (percentage inhibition at 0.25 mg/mL and, respectively 0.5 mg/mL concentration): Acremonium strictum (76.88%; 100%) Aspergillus flavus (15.38%; 60.51%) |
|||||
Antibacterial activity (R=6-OCH | 3 | , 7-Cl) Staphylococcus aureus (MIC = 1.25 mg/mL) Bacillus subtilis (MIC = 1.25 mg/mL) Klebsiella pneumoniae (MIC = 0.625 mg/mL) Anti biofilm activity (R=6-OCH3, 100 μg/mL) Antifungal activity (R=7-Cl): Candida albicans (MIC = 0.156 mg/mL) |
|||||
|
14 |
|
Antibacterial activity Mycobacterium tuberculosis H37Rv (MIC = 6.25 µg/mL) |
|||||
|
15 |
|
Antiviral activity | |||||
[ | |||||||
] | [ | 44] |
|||||
|
17 |
|
|
|||||
|
18 |
|
|
|||||
|
19 |
|
|
|||||
|
20 |
|
|
|||||
|
21 |
|
|
|||||
Human cytomegalovirus | (EC 50 = 0.126 nM) |
||||||
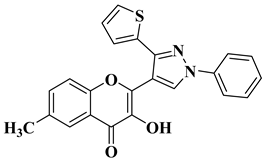
|
16 |
|
Antiviral activity Chikungunya Virus (IC50 = 0.44 µM) |
|||||
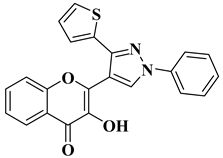
|
17 |
|
Antiviral activity Chikungunya Virus (IC50 = 0.45 µM) |
22 |
|
|
||
|
23 |
|
|
|||||
|
24 |
|
|
|||||
|
25 |
|
|
|||||
|
26 |
|
|
|||||
|
27 |
|
|
|||||
|
28 |
|
|
References
- Berim, A.; Gang, D.R. Methoxylated flavones: Occurrence, importance, biosynthesis. Phytochem. Rev. 2016, 15, 363–390.
- Ververidis, F.; Trantas, E.; Douglas, C.; Vollmer, G.; Kretzschmar, G.; Panopoulos, N. Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2007, 2, 1214–1234.
- Hou, D.X.; Kumamoto, T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: Direct binding and molecular modeling. Antioxid. Redox Signal. 2010, 13, 691–719.
- Polier, G.; Ding, J.; Konkimalla, B.V.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011, 2, e182.
- Golub, A.G.; Bdzhola, V.G.; Ostrynska, O.V.; Kyshenia, I.V.; Sapelkin, V.M.; Prykhod’ko, A.O.; Kukharenko, O.P.; Yarmoluk, S.M. Discovery and characterization of synthetic 4′-hydroxyflavones—New CK2 inhibitors from flavone family. Bioorg. Med. Chem. 2013, 21, 6681–6689.
- Chao, S.W.; Su, M.Y.; Chiou, L.C.; Chen, L.C.; Chang, C.I.; Huang, W.J. Total Synthesis of Hispidulin and the Structural Basis for Its Inhibition of Proto-oncogene Kinase Pim-1. J. Nat. Prod. 2015, 78, 1969–1976.
- Sedlacek, H.; Czech, J.; Naik, R.; Kaur, G.; Worland, P.; Losiewicz, M.; Parker, B.; Carlson, B.; Smith, A.; Senderowicz, A.; et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996, 9, 1143–1168.
- Pontes, O.; Costa, M.; Santos, F.; Sampaio-Marques, B.; Dias, T.; Ludovico, P.; Baltazar, F.; Proença, F. Exploitation of new chalcones and 4H-chromenes as agents for cancer treatment. Eur. J. Med. Chem. 2018, 157, 101–114.
- Monasterio, A.; Urdaci, M.; Pinchuk, I.; Moratalla, N.; Martínez, I. Flavonoids induce apoptosis in human leukemia U937 cells through caspase-and caspase-calpain-dependent pathways. Nutr. Cancer 2004, 50, 90–100.
- Moreira, J.; Ribeiro, D.; Silva, P.M.A.; Nazareth, N.; Monteiro, M.; Palmeira, A.; Saraiva, L.; Pinto, M.; Bousbaa, H.; Cidade, H. New alkoxy flavone derivatives targeting caspases: Synthesis and antitumor activity evaluation. Molecules 2019, 24, 129.
- Kariagina, A.; Doseff, A.I. Anti-Inflammatory Mechanisms of Dietary Flavones: Tapping into Nature to Control Chronic Inflammation in Obesity and Cancer. Int. J. Mol. Sci. 2022, 23, 15753.
- Occhiuto, C.J.; Moerland, J.A.; Leal, A.S.; Gallo, K.A.; Liby, K.T. The Multi-Faceted Consequences of NRF2 Activation throughout Carcinogenesis. Mol. Cells 2023, 46, 176–186.
- Ross, J.A.; Kasum, C.M. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu. Rev. Nutr. 2002, 22, 19–34.
- Jiang, M.; Zhu, M.; Wang, L.; Yu, S. Anti-tumor effects and associated molecular mechanisms of myricetin. Biomed. Pharmacother. 2019, 120, 109506.
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A dietary molecule with diverse biological activities. Nutrients 2016, 8, 90.
- Zang, W.; Wang, T.; Wang, Y.; Li, M.; Xuan, X.; Ma, Y.; Zhao, G. Myricetin exerts anti-proliferative, anti-invasive, and pro-apoptotic effects on esophageal carcinoma EC9706 and KYSE30 cells via RSK2. Tumor Biol. 2014, 35, 12583–12592.
- Chen, Y.C.; He, X.L.; Qi, L.; Shi, W.; Yuan, L.-W.; Huang, M.-Y.; Xu, Y.-L.; Chen, X.; Gu, L.; Zhang, L.-L.; et al. Myricetin inhibits interferon-γ-induced PD-L1 and IDO1 expression in lung cancer cells. Biochem. Pharmacol. 2022, 197, 114940.
- Han, S.-H.; Lee, J.-H.; Woo, J.-S.; Jung, G.-H.; Jung, S.-H.; Han, E.-J.; Kim, B.; Cho, S.-D.; Nam, J.-S.; Che, J.H.; et al. Myricetin induces apoptosis and autophagy in human gastric cancer cells through inhibition of the PI3K/Akt/mTOR pathway. Heliyon 2022, 8, e09309.
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273.
- Li, L.; Ma, H.; Li, D.; Shu, Q.; Wang, T.; Song, X.; Xu, H. Myricetin alleviates the formaldehyde-enhanced Warburg effect in tumor cells through inhibition of HIF-1α. Toxicology and Appl. Pharmacol. 2022, 454, 116246.
- Gu, L.; Li, Z.; Zhang, X.; Chen, M.; Zhang, X. Identification of MAP Kinase Kinase 3 as a protein target of myricetin in non-small cell lung cancer cells. Biomed. Pharmacother. 2023, 161, 114460.
- El Menyiy, N.; Aboulaghras, S.; Bakrim, S.; Moubachir, R.; Taha, D.; Khalid, A.; Abdalla, A.N.; Algarni, A.S.; Hermansyah, A.; Ming, L.C.; et al. Genkwanin: An emerging natural compound with multifaceted pharmacological effects. Biomed. Pharmacother. 2023, 165, 115159.
- Li, Y.; Hong, J.; Li, H.; Qi, X.; Guo, Y.; Han, M.; Wang, X. Genkwanin nanosuspensions: A novel and potential antitumor drug in breast carcinoma therapy. Drug Deliv. 2017, 24, 1491–1500.
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants 2020, 9, 461.
- Grigalius, I.; Petrikaite, V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules 2017, 22, 2169.
- Zhao, L.; Yuan, X.; Wang, J.; Feng, Y.; Ji, F.; Li, Z.; Bian, J. A review on flavones targeting serine/threonine protein kinases for potential anticancer drugs. Bioorg. Med. Chem. 2019, 27, 677–685.
- Chao, S.H.; Price, D.H. Flavopiridol Inactivates P-TEFb and Blocks Most RNA Polymerase II Transcription in Vivo. J. Biol. Chem. 2001, 276, 31793–31799.
- Hassan, A.H.; Choi, E.; Yoon, Y.M.; Lee, K.W.; Yoo, S.Y.; Cho, M.C.; Yang, J.S.; Kim, H.I.; Hong, J.Y.; Shin, J.-S.; et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur. J. Med. Chem. 2019, 161, 559–580.
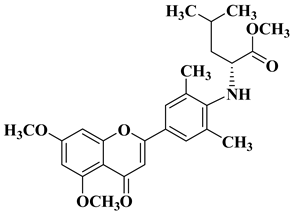
- Hassan, A.H.E.; Lee, K.T.; Lee, Y.S. Flavone-based arylamides as potential anticancers: Design, synthesis and in vitro cell-based/cell-free evaluations. Eur. J. Med. Chem. 2020, 187, 111965.
- Li, X.; Zhang, C.; Guo, S.; Rajaram, P.; Lee, M.; Chen, G.; Fong, R.; Gonzalez, A.; Zhang, Q.; Zheng, S.; et al. Structure-activity relationship and pharmacokinetic studies of 3-O-substitutedflavonols as anti-prostate cancer agents. Eur. J. Med. Chem. 2018, 157, 978–993.
- Németh-Rieder, A.; Keglevich, P.; Hunyadi, A.; Latif, A.D.; Zupkó, I.; Hazai, L. Synthesis and In Vitro Anticancer Evaluation of Flavone—1,2,3-Triazole Hybrids. Molecules 2023, 28, 626.
- Mobbili, G.; Romaldi, B.; Sabbatini, G.; Amici, A.; Marcaccio, M.; Galeazzi, R.; Laudadio, E.; Armeni, T.; Minnelli, C. Identification of Flavone Derivative Displaying a 4-Aminophenoxy Moiety as Potential Selective Anticancer Agent in NSCLC Tumor Cells. Molecules 2023, 28, 3239.
- Bollikolla, H.B.; Anandam, R.; Chinnam, S.; Varala, R.; Khandapu, B.M.K.; Kapavarapu, R.; Syed, K.S.; Dubasi, N.; Syed, M.A. C-Dimethylated Flavones as Possible Potential Anti-Tubercular and Anticancer Agents. Chem. Biodivers. 2023, 20, e202201201.
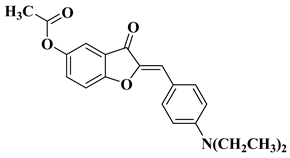
- Schoepfer, J.; Fretz, H.; Chaudhuri, B.; Muller, L.; Seeber, E.; Meijer, L.; Lozach, O.; Vangrevelinghe, E.; Furet, P. Structure-based design and synthesis of 2-benzylidene-benzofuran-3-ones as flavopiridol mimics. J. Med. Chem. 2002, 45, 1741–1747.
- Priyadarshani, G.; Nayak, A.; Amrutkar, S.M.; Das, S.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C. Scaffold-Hopping of Aurones: 2-Arylideneimidazopyridinones as Topoisomerase IIα-Inhibiting Anticancer Agents. ACS Med. Chem. Lett. 2016, 7, 1056–1061.
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969.
- Lawrence, N.J.; Rennison, D.; McGown, A.T.; Hadfield, J.A. The total synthesis of an aurone isolated from Uvaria hamiltonii: Aurones and flavones as anticancer agents. Bioorg. Med. Chem. Lett. 2003, 13, 3759–3763.
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorg. Med. Chem. 2009, 17, 8073–8085.
- Hadjeri, M.; Barbier, M.; Ronot, X.; Mariotte, A.M.; Boumendjel, A.; Boutonnat, J. Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J. Med. Chem. 2003, 46, 2125–2131.
- Sim, H.M.; Wu, C.P.; Ambudkar, S.V.; Go, M.L. In vitro and in vivo modulation of ABCG2 by functionalized aurones and structurally related analogs. Biochem. Pharmacol. 2011, 82, 1562–1571.
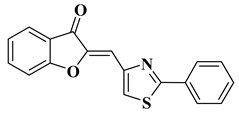
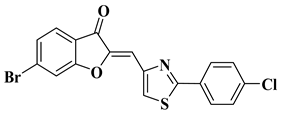
- Coman, F.M.; Mbaveng, A.T.; Marc, G.; Leonte, D.; Brém, B.; Vlase, L.; Imre, S.; Kuete, V.; Zaharia, V. Heterocycles 47. Synthesis, Characterization and Biological Evaluation of some New Thiazole Aurones as Antiproliferative Agents. Farmacia 2020, 68, 492–506.
- Semenov, I.; Akyuz, C.; Roginskaya, V.; Chauhan, D.; Corey, S.J. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk. Res. 2002, 26, 271–280.
- Jeon, K.-H.; Park, S.; Shin, J.-H.; Jung, A.-R.; Hwang, S.-Y.; Seo, S.H.; Jo, H.; Na, Y.; Kwon, Y. Synthesis and evaluation of 7-(3-aminopropyloxy)-substituted flavone analogue as a topoisomerase IIα catalytic inhibitor and its sensitizing effect to enzalutamide in castration-resistant prostate cancer cells. Eur. J. Med. Chem. 2023, 246, 114999.
- Su, L.; Li, W.; Liu, K.; Wang, Q. Synthesis and anti-proliferative activities of 5,6,7-trimethoxyflavones and their derivatives. Nat. Prod. Res. 2022, 36, 4070–4075.
- Zhang, N.; Yang, J.; Li, K.; Luo, J.; Yang, S.; Song, J.-R.; Chen, C.; Pan, W.-D. Synthesis of flavone derivatives via N-amination and evaluation of their anticancer activities. Molecules 2019, 24, 2723.
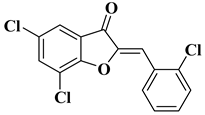
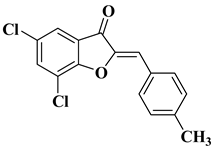
- Elhadi, A.A.; Osman, H.; Iqbal, M.A.; Rajeswari, S.K.; Ahamed, M.B.K.; Majid, A.M.A.; Rosli, M.M.; Razak, I.A.; Majid, A.S.A. Synthesis and structural elucidation of two new series of aurone derivatives as potent inhibitors against the proliferation of human cancer cells. Med. Chem. Res. 2015, 24, 3504–3515.
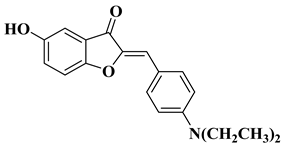
- Cheng, H.; Zhang, L.; Liu, Y.; Chen, S.; Cheng, H.; Lu, X.; Zheng, Z.; Zhou, G.-C. Design, synthesis and discovery of 5-hydroxyaurone derivatives as growth inhibitors against HUVEC and some cancer cell lines. Eur. J. Med. Chem. 2010, 45, 5950–5957.
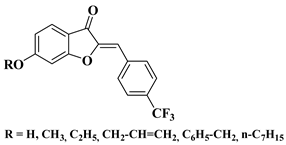
- Zheng, X.; Wang, H.; Liu, Y.-M.; Yao, X.; Tong, M.; Wang, Y.-H.; Liao, D.-F. Synthesis, Characterization, and Anticancer Effect of Trifluoromethylated Aurone Derivatives. J. Heterocycl. Chem. 2014, 6, 1098–1107.
- Uesawa, Y.; Sakagami, H.; Ikezoe, N.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative structure-cytotoxicity relationship of aurones. Anticancer Res. 2017, 37, 6169–6176.
- Demirayak, S.; Yurttas, L.; Gundogdu-Karaburun, N.; Karaburun, A.C.; Kayagil, I. Synthesis and anticancer activity evaluation of new aurone derivatives. J. Enzym. Inhib. Med. Chem. 2015, 30, 816–825.
- Lathwal, E.; Kumar, S.; Sahoo, P.K.; Ghosh, S.; Mahata, S.; Nasare, V.D.; Kumar, S. Synthesis, cytotoxic evaluation and structure activity relationship of pyrazole hybrid aurones on gastric cancer (AGS) cell lines. Results Chem. 2022, 4, 100590.
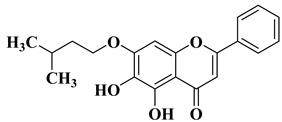
- Gao, L.; Tang, Z.; Li, T.; Wang, J. Myricetin exerts anti-biofilm activity and attenuates osteomyelitis by inhibiting the TLR2/MAPK pathway in experimental mice. Microb. Pathog. 2023, 182, 106165.
- Ashok, D.; Kifah, M.A.; Lakshmi, B.V.; Sarasija, M.; Adam, S. Microwave-assisted one-pot synthesis of some new flavonols by modified Algar–Flynn–Oyamada reaction and their antimicrobial activity. Chem. Heterocycl. Compd. 2016, 52, 172–176.
- Lv, X.H.; Liu, H.; Ren, Z.L.; Wang, W.; Tang, F.; Cao, H.Q. Design, synthesis and biological evaluation of novel flavone Mannich base derivatives as potential antibacterial agents. Mol. Divers. 2019, 23, 299–306.
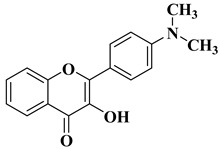
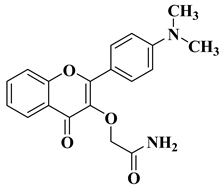
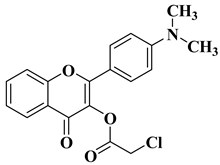
- Khdera, H.A.; Saad, S.Y.; Moustapha, A.; Kandil, F. Synthesis of new flavonoid derivatives based on 3-hydroxy-4′-dimethylamino flavone and study the activity of some of them as antifungal. Heliyon 2022, 8, e12062.
- Kumar, G.; Lathwal, E.; Saroha, B.; Kumar, S.; Kumar, S.; Chauhan, N.S.; Kumar, T. Synthesis and Biological Evaluation of Quinoline-Based Novel Aurones. ChemistrySelect 2020, 5, 3539–3543.
- Pan, H.; He, J.; Yang, Z.; Yao, X.; Zhang, H.; Li, R.; Xiao, Y.; Zhao, C.; Jiang, H.; Liu, Y.; et al. Myricetin possesses the potency against SARS-CoV-2 infection through blocking viral-entry facilitators and suppressing inflammation in rats and mice. Phytomedicine 2023, 116, 154858.
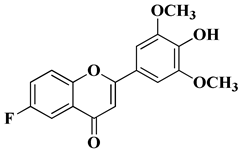
- Fujimoto, K.J.; Nema, D.; Ninomiya, M.; Koketsu, M.; Sadanari, H.; Takemoto, M.; Daikoku, T.; Murayama, T. An in silico-designed flavone derivative, 6-fluoro-4′-hydroxy-3′,5′-dimetoxyflavone, has a greater anti-human cytomegalovirus effect than ganciclovir in infected cells. Antivir. Res. 2018, 154, 10–16.
- Badavath, V.N.; Jadav, S.; Pastorino, B.; de Lamballerie, X.; Sinha, N.B.; Jayaprakash, V. Synthesis and Antiviral Activity of 2-aryl-4H-chromen-4-one Derivatives Against Chikungunya Virus. Lett. Drug Des. Discov. 2016, 13, 1019–1024.