Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jeong Tae Do | -- | 3106 | 2023-05-23 10:00:52 | | | |
| 2 | Catherine Yang | Meta information modification | 3106 | 2023-05-23 10:40:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yoon, S.H.; Kim, G.Y.; Choi, G.T.; Do, J.T. Organ Abnormalities Caused by Turner Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/44696 (accessed on 09 August 2026).
Yoon SH, Kim GY, Choi GT, Do JT. Organ Abnormalities Caused by Turner Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/44696. Accessed August 09, 2026.
Yoon, Sang Hoon, Ga Yeon Kim, Gyu Tae Choi, Jeong Tae Do. "Organ Abnormalities Caused by Turner Syndrome" Encyclopedia, https://encyclopedia.pub/entry/44696 (accessed August 09, 2026).
Yoon, S.H., Kim, G.Y., Choi, G.T., & Do, J.T. (2023, May 23). Organ Abnormalities Caused by Turner Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/44696
Yoon, Sang Hoon, et al. "Organ Abnormalities Caused by Turner Syndrome." Encyclopedia. Web. 23 May, 2023.
Copy Citation
Turner syndrome (TS), a genetic disorder due to incomplete dosage compensation of X-linked genes, affects multiple organ systems, leading to hypogonadotropic hypogonadism, short stature, cardiovascular and vascular abnormalities, liver disease, renal abnormalities, brain abnormalities, and skeletal problems. Patients with TS experience premature ovarian failure with a rapid decline in ovarian function caused by germ cell depletion, and pregnancies carry a high risk of adverse maternal and fetal outcomes. Aortic abnormalities, heart defects, obesity, hypertension, and liver abnormalities, such as steatosis, steatohepatitis, biliary involvement, liver cirrhosis, and nodular regenerative hyperplasia, are commonly observed in patients with TS.
Turner syndrome
X monosomy
X chromosome inactivation
organ abnormalities
1. Introduction
Turner syndrome (TS) is one of the most common disorders caused by chromosomal abnormalities, affecting approximately 1 in 2500 live female births. It is the only viable monosomy syndrome caused by partial or complete loss of one of the two sex chromosomes [1]. TS was first reported in 1938 by Henry H. Turner as a syndrome of infantilism, congenital webbed neck, and cubitus valgus, and Ford et al. found that the disease was caused by sex chromosomal abnormality in 1959 [2][3]. The most common karyotype in TS is 45,X, accounting for 40–50% of all cases of TS, whereas 45,X/46,XX or 45,X/47,XXX mosaicism account for 20–30%. The remaining cases include Y chromosome variants and X chromosome structural abnormalities, such as isochromosome Xq, deletion of Xp or Xq (which can occur as mosaicism), and ring X (which is always mosaic) [4] (Figure 1). Thus, in TS, only one X chromosome is normal and the others are absent or abnormal. The diagnosis of TS has traditionally relied on the clinical phenotype in addition to standard chromosomal analysis [5]. Total or partial loss of one of the two sex chromosomes affects biological pathways and networks [5], and, in some cases, SHOX gene defects have been linked to certain phenotypes of TS [6][7] (Table 1).
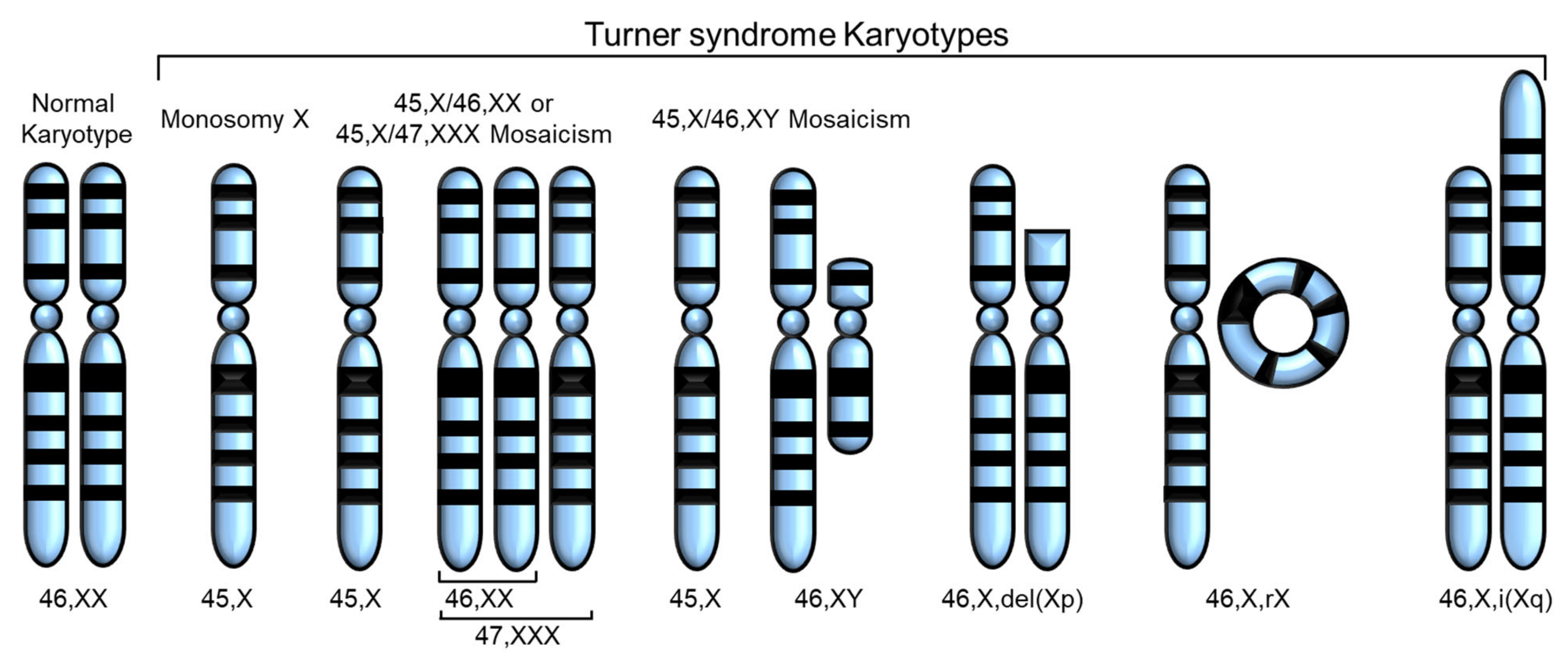
Figure 1. Various karyotypes of Turner syndrome (TS) modified from Huang et al. [8]. Partial or complete loss of the second sex chromosome results in TS. The most common karyotype in TS is monosomy X (45,X), and the others are 45,X/46,XX or 45,X/47,XXX mosaicism, 45,X/45,XY mosaicism, deletion of Xp or Xq, ring X (46,X,rX), and isochromosome Xq.
Table 1. Genes associated with Turner Syndrome (TS).
| Gene | Location | Expression in TS | Associated Phenotype | Reference |
|---|---|---|---|---|
| SHOX | Xp22.33 and Y chromosome (PAR1) | Decreased expression | Short stature, Madelung wrist deformity, Intellectual disabilities |
[9][10][11][12][13] |
| ARSD, ARSE, ARSF | Xp22.3 | Loss owing to contiguous gene deletion syndrome | Chondrodysplasia punctata | [14][15][16] |
| STS | Xp22.31 | Loss owing to contiguous gene deletion syndrome | X-linked ichthyosis | [13][14][15][16] |
| GPR143 | Xp22.2 | Loss owing to contiguous gene deletion syndrome | Ocular albinism type I | [14][15][16] |
| ANOS1 | Xp23.3 | Loss owing to contiguous gene deletion syndrome | Kallmann syndrome | [14][15][16] |
| RPS4X | Xq13.1 | Downregulation | N/A | [13][17][18][19] |
| CD99 | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20] |
| CSF2RA | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20][21][22] |
| MYL9 | 20q11.23 | Downregulated | N/A | [20] |
| MYLPF | 16p11.2 | Downregulated | N/A | [20] |
| IGFBP2 | 2q35 | Downregulated | N/A | [20] |
Individuals with TS are at an increased risk of endocrine diagnoses, including diabetes, thyroid and parathyroid disorders, celiac disease, and osteoporosis [23][24], as well as cardiovascular diseases, including arrhythmia, ischemic heart disease, hypertension, hyperlipidemia, and stroke. This is supported by the increased use of prescription drugs by patients with TS [25]. The 45,X karyotype is associated with the highest rates of morbidity and mortality, whereas the mosaic karyotype is associated with a low prevalence for cardiovascular, metabolic, renal, and reproductive phenotypes [26][27][28][29][30][31][32][33]. Despite ongoing research, no feasible treatment has been proposed owing to the severe effects of losing an entire chromosome and the numerous genes that are simultaneously affected [34].
2. Fertility Problems
Infertility is one of the most common symptoms of TS, despite low rates of spontaneous pregnancies [31][35][36][37]. TS is accompanied by hypogonadotropic hypogonadism in almost all patients, leading to primary or secondary amenorrhea and infertility owing to premature ovarian failure (POF) (affecting approximately 95% of women with TS) and premature ovarian insufficiency [28][38][39]. Therefore, women with TS do not produce enough eggs or the necessary hormones to support pregnancy. The ovaries in a 45,X fetus appear to develop normally until birth; however, follicular atresia is induced by birth or early childhood [40]. Moreover, 5–20% of girls with TS retain enough follicles to permit spontaneous menarche, even if early menopause typically follows. Women with TS who have a mosaic karyotype, or experience spontaneous puberty, have follicles in one or both ovaries [39]. Furthermore, those with low levels of 45,X/46,XX mosaicism are less severely affected and have a high likelihood of experiencing spontaneous menstruation and pregnancy, although karyotype does not always predict phenotype [41][42][43]. Accelerated germ cell death is presumed to be the major mechanism causing germ cell depletion in patients with TS.
Reynaud et al. analyzed 10 aborted fetuses with TS and found that the number of germ cells in the genital ridge was similar to that in the control group up to 12 weeks of gestation, indicating normal migration of primordial germ cells in fetuses with TS [44]. However, differences were observed from 18 weeks of gestation, where germ cells were rarely detected, and completely absent at 25 weeks of gestation in fetuses with 45,X TS. Moreover, primordial and antral follicles were absent in fetuses with 45,X TS, although they were present in fetuses with TS with mosaicism. These studies suggest that folliculogenesis is severely impaired in ovaries of patients with TS, possibly owing to the loss of germ cells [44]. Additionally, the eggs from women with TS might be of poor quality, decreasing the chances of successful fertilization and pregnancy [45].
TS can also cause abnormalities in the structure and function of the uterus, affecting the implantation and growth of fertilized eggs [46]. Only about a quarter of people with TS have a fully developed uterus in size and shape, while most others have a slightly smaller uterus; about one-third have an immature form of the uterus. Notably, the difference in the size of the uterus between women with TS and those with a normal karyotype is not significant; however, on average, women with TS have a smaller uterine volume than those with a normal karyotype. The size of the uterus in individuals with TS can be influenced by various factors, including the age of the patient, duration of estrogen use, use of hormone replacement therapy (HRT), and type of estrogen medication administered. However, with appropriate and timely treatment, women with TS can achieve normal uterine development [46].
In addition, an imbalance in sex hormone levels affects the fertility of patients with TS. Women with TS showed 30–50% lower levels of androgens, including testosterone, free androgen index, androstenedione, and dehydroepiandrosterone sulfate, than those with a normal karyotype, but an increase in Follicle stimulating hormone (FSH), Luteinizing hormone (LH), and estrone sulfate levels up to twice the normal range [47]. High levels of FSH and LH during adolescence are linked to reduced ovarian function [48]. However, patients with TS showed a normal biphasic age pattern of reproductive hormones, with peak FSH and LH levels occurring at three months of age, followed by a subsequent decrease to minimal levels during mid-childhood and reactivation at puberty [48][49].
Pregnancy is rare among patients with TS and shows a high risk of miscarriage, stillbirth, and birth defects [50]. Only 2–5% of patients with TS become pregnant spontaneously, and approximately 3.8% of patients with TS have one or more live-born children [28][35]. Both natural and medically assisted pregnancies in patients with TS have a higher risk of adverse maternal and fetal outcomes than those in healthy women. For instance, 23–50% of women with TS have congenital heart disease, and pregnancy causes a 50% increase in cardiac output, making patients with TS susceptible to aortic dissection or rupture. As a result, the risk of death during pregnancy for patients with TS can reach up to 2% [51][52].
3. Heart and Cardiovascular Disease
Congenital and acquired heart defects and cardiovascular conditions are the leading cause of death in patients with TS, affecting about 25–50% of cases, with a higher incidence in those with 45,X karyotypes than in those with other TS variants [53]. Miyabara et al. conducted an autopsy of a 20-week-old fetus with 45,X karyotype and found that the wall of the aortic arch was much thinner than normal and that the number of smooth muscle cells and elastic fibers in the aorta was significantly reduced [54]. Anomalies of the coronary arteries are diverse and include many variants other than two arteries originating from aortic sinuses [55]. Many types of coronary artery anomalies have been reported in TS, especially in patients with bicuspid aortic valve (BAV) [56][57][58]. Although not all patients with TS have arch anomalies, aortic arch anomalies are common in TS owing to the complex embryological development of this vessel [59][60][61]. The most common anomalies include elongation of the arch and aberrant right subclavian artery [62][63]. Patients with TS and aortic arch anomalies are also at risk of developing aortic dilation, which could increase the risk of aortic dissection, occurring in 1–2% of patients with TS [64][65].
Aortic arch hypoplasia is another congenital aortic anomaly associated with TS and may vary in severity from mild aortic stenosis to severe transverse arch hypoplasia, interrupted aortic arch, or hypoplastic left heart syndrome [66]. Patients with TS are also prone to increased carotid artery thickness and arterial diameter, possibly owing to estrogen deficiency, which can be attenuated by estrogen hormone therapy [67][68][69]. Abnormalities of the venous system, such as hypoplasia of the portal vein system, are also observed in patients with TS, and vascular atrophy is involved in liver dysfunction [70].
As abnormal extracellular matrix (ECM) composition induces aortic structural malformation, matrix metalloproteinases (MMPs, a degradation factor of ECM), and tissue inhibitors of matrix metalloproteinases (TIMPs, inhibitor of MMPs) are involved in aortic abnormalities [71]. Increased expression of MMPs and reduced expression of TIMP1 and TIMP3 can lead to the degradation of ECM components of the aortic wall, resulting in thinning of the aortic wall and enlargement of the diameter. These changes are implicated in the pathogenesis of various abnormal aortic morphogeneses, such as BAV and aortic aneurysms [71][72]. Therefore, hemizygous expression of TIMP1 on the Xp locus in patients with TS may increase susceptibility to abnormal aortic morphogenesis. Decreased expression of TIMP3, a TIMP1 paralogue on chromosome 22, can augment the risk for aortopathy and BAV [71]. In addition, TIMP1 is hypermethylated, which suggests that this gene is epigenetically inactive in patients with TS [73]. Moreover, reduced expression of TIMP1 and TIMP3 was observed in the euploid population with BAV and aortopathy [72].
4. Liver Abnormalities
Although liver involvement is mostly asymptomatic in patients with TS, a wide range of abnormal phenotypes may be observed in the liver, including steatosis, steatohepatitis, liver cirrhosis, biliary involvement, and nodular regenerative hyperplasia (NRH) [74][75][76][77][78][79]. Singh et al. reported that approximately twice the number of girls with TS showed liver enzyme elevation (alanine aminotransferase and aspartate aminotransferase) compared with normal controls [80]. These liver enzyme levels have clinical significance as girls with TS with elevated liver enzyme levels are more likely to be diagnosed with liver disease [80]. For example, hypertransaminasemia is common in patients with TS and is typically associated with hepatic steatosis, which can also be caused by other factors, such as diabetes mellitus and dyslipidemia [81]. In addition, women with TS with elevated liver enzymes are overweight and exhibit high levels of cholesterol, triglycerides, apolipoproteins A and B, and gamma-glutamyl transferase [82]. Excessive body weight is a common cause of liver disease in patients with TS [82][83]. Patients who are overweight (>25 kg/m2), as defined by body mass index (BMI) values, frequently experience insulin secretion disorders and diabetes mellitus. However, increased weight and BMI in patients with TS are not necessarily estrogen-related. Moreover, the lack of estrogen or GH (Growth Hormone) treatment is not the primary cause of the increase in liver enzymes.
5. Kidney Abnormalities
Kidney abnormalities are common in patients with TS, with a prevalence of 33–70%, and include kidney and urinary tract anomalies, such as abnormal ureter structure leading to urine regurgitation, horseshoe kidney (kidney fusion), renal aplasia, duplex collecting system, single unilateral kidney, and formation of cilia and cysts in the kidney [84][85][86][87]. The most frequently reported renal anomaly is the horseshoe kidney, which occurs in 20–45% of patients with TS, whereas it is observed in less than 3% of the general population [88][89]. Horseshoe kidney is caused by the fusion of the two kidneys, forming a U-shaped structure. While patients with TS with horseshoe kidney may be asymptomatic during childhood, they may experience recurrent urinary tract infections and kidney stones in the later stages of the condition [85][90][91][92]. The incidence of renal malformations is significantly higher in patients with TS with a non-mosaic 45,X karyotype than in those with mosaicism, probably owing to lymphatic retention and organ system compression [91]. Hypertension can also be caused by renal malformations besides aortic stenosis and intrarenal vascular changes in patients with TS [91]. Other rare cases of malformations include rotation and postural abnormalities, severe and mild hydronephrosis, and unilateral/bilateral overlap collector type.
In TS, congenital anomalies of the kidney and renal-urinary tract (CAKUT) can manifest as hemiplegic, neoplastic, and polycystic kidneys. While most patients with horseshoe kidneys have normal kidney function, renal hypoplasia may lead to impaired renal function. X-structural abnormalities were observed in 68.7% of patients with a non-mosaic 45,X karyotype and in 9.0% of patients with a 45,X mosaic karyotype. In those with 45,X monosomy, 45,X with mosaicism, and X-structural abnormalities, the CAKUT incidence was 11.5%, 7.4%, and 25.0%, respectively, indicating a reduced ability to form kidneys with non-mosaic X chromosome abnormalities [85].
6. Skeletal Abnormalities and Short Stature
Girls with TS often suffer from reduced bone density and delayed bone formation owing to estrogen deficiency during adolescence [93][94]. Osteopenia or osteoporosis are identified as common factors for problems in bone formation [93][95][96][97][98]. Growth retardation of the joints of the finger bones was also noticeable in patients with TS compared with normal controls. This difference in bone formation between patients with TS and normal controls is minor until the age of 10 years, but becomes more significant during puberty [99].
Bone density analysis showed decreased bone density in patients with TS in various areas [100][101]. For example, bone mineral apparent density (BMAD) in patients with TS was significantly lower in the femoral neck—an area of predominantly cortical bone—than in normal controls [100][102]. In addition, proximal radius and cortical volumetric bone mineral density (vBMD) exhibited a decreasing trend in cortical thickness [100][103]. BMD was maintained predominantly in trabecular bone, and BMD in the lumbar spine, an area rich in trabecular bone, was not significantly different between TS and control groups [100]. Women with TS also showed low BMAD in the cortical and trabecular bone of the forearm [104], and the width of the ultradistal radius (predominantly in the trabecular bone) was reduced [102].
7. Brain Abnormalities
While no visible brain abnormalities are apparent, structural, electrophysiological, cognitive, and psychosocial studies have reported differences between patients with TS and normal control. Reiss et al. suggested that the brain structure of women with TS could be distinguishable from that of age-matched controls [105]. Several other studies also suggested that patients with TS had a small volume of cerebral hemispheres and an increased volume of cerebrospinal fluid and the fourth ventricle [106][107]. The size of gray and white matter mainly determines brain volume and size. When comparing patients with TS against controls, although not statistically significant, there was an increase in gray matter in the right superior temporal gyrus and left amygdala and an increase in white matter in the left superior temporal gyrus [108]. However, many reports also suggested that the brain structure was smaller than that of the control group. Compared with that of controls, in individuals with TS, there is a reduction in gray matter in various regions, including the right calcarine cortex, precentral region, supramarginal gyri, cuneus, lingual cortex, superior parietal, rostral anterior portion, pericalcarine, and postcentral and precuneus of the right hemisphere’s cingulate cortex, as well as a reduction in white matter in the entorhinal cortex, pars opercularis, frontal pole, and occipital lobe [109][110][111]. Most gray matter reductions are related to surface area reduction [109].
8. Relevance to X Chromosome Inactivation and Escape Genes
During the early development of mammals, one of the two X chromosomes in females (XX) is randomly inactivated by X chromosome inactivation (XCI), by which the total amount of X-linked genes expressed in females becomes equivalent to that in males (XY) [112][113]. Normal female somatic cells contain one active X chromosome (Xa) and one inactive X chromosome (Xi), resulting in a XaXi state. If X-linked genes in the Xi are completely silenced, the removal of Xi from XaXi may not have a harmful effect on cells. However, in TS the complete or partial loss of the Xi leads to a myriad of abnormalities. This is because some genes located outside the condensed heterochromatin of the Xi can escape from inactivation and be expressed, leading to differences in the number of expressed X-linked genes between 46,XaXi and 45,Xa states. These genes that are expressed from the Xi are called escape genes [114][115]. Approximately 15% and 3% of X-linked genes in humans and mice, respectively, are escape genes [115]. Human X chromosomes have pseudoautosomal regions (PARs) that behave like autosomes where crossing over strictly occurs (Table 1). Genes within PARs on the X chromosome usually escape from XCI [116]. The PAR1 genes, including SHOX, play essential roles in the phenotypic traits associated with TS, including short stature, Madelung’s wrist deformity, and intellectual disabilities [9][10]. Variations in the expression of these genes may contribute to growth deficits or increased height in affected individuals. Decreased expression of SHOX contributes to growth deficits observed in patients with TS, whereas increased expression in Klinefelter syndrome (47,XXY), Triple X (47,XXX), and Double Y (47,XYY) is associated with increased height [11].
In addition, 12 genes (AKAP17A, ASMT, ASMTL, CD99, CD99P1, CRLF2, CSF2RA, DHRSX, FABP5P13, GTPBP6, IL3RA, PLCXD1, PPP2R3B, P2RY8, SHOX, SLC25A6, XG, and ZBED1) located outside the PAR1 region have a single functionally Y homolog and are broadly expressed in human tissues [12]. USP9X genes on the X chromosome could evade Xi and be expressed in both human adult and embryonic tissues [117]. Quilter et al. found that the expression of escape genes, USP9X and ZFX, was associated with immune cell development, oocyte growth, and ovarian development [118].
Variants of the KDM6A gene, known to escape XCI, are also associated with Kabuki syndrome, a multisystem syndrome with TS-like phenotypic traits, such as growth delay, short stature, varying degrees of intellectual disabilities, skeletal and renal abnormalities, and congenital heart defects [119][120]. RPS4X and RSPS4Y are also considered dosage-sensitive genes, and several studies reported RPS4X downregulation in TS [17][18][19]. Wang et al. identified 25 upregulated and 60 downregulated genes in patients with TS compared with those in normal women and found five genes, including CD99, CSF2RA, MYL9, MYLPF, and IGFBP2, possibly involved in the pathogenesis of TS [20]. In addition, epigenetic mechanisms, such as DNA methylation, are also involved in the etiology of TS [121]. However, further studies are required to understand the correlation between escape genes and TS.
References
- Saenger, P. Turner’s syndrome. N. Engl. J. Med. 1996, 335, 1749–1754.
- Turner, H.H. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938, 23, 566–574.
- Ford, C.E.; Jones, K.W.; Polani, P.E.; De Almeida, J.C.; Briggs, J.H. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner’s syndrome). Lancet 1959, 1, 711–713.
- Bollig, K.J.; Mainigi, M.; Senapati, S.; Lin, A.E.; Levitsky, L.L.; Bamba, V. Turner syndrome: Fertility counselling in childhood and through the reproductive lifespan. Curr. Opin. Endocrinol. Diabetes Obes. 2023, 30, 16–26.
- Gravholt, C.H.; Viuff, M.; Just, J.; Sandahl, K.; Brun, S.; van der Velden, J.; Andersen, N.H.; Skakkebaek, A. The Changing Face of Turner Syndrome. Endocr. Rev. 2023, 44, 33–69.
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 1997, 16, 54–63.
- Ellison, J.W.; Wardak, Z.; Young, M.F.; Gehron Robey, P.; Laig-Webster, M.; Chiong, W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum. Mol. Genet. 1997, 6, 1341–1347.
- Huang, A.C.; Olson, S.B.; Maslen, C.L. A Review of Recent Developments in Turner Syndrome Research. J. Cardiovasc. Dev. Dis. 2021, 8, 138.
- Binder, G.; Fritsch, H.; Schweizer, R.; Ranke, M.B. Radiological signs of Leri-Weill dyschondrosteosis in Turner syndrome. Horm. Res. 2001, 55, 71–76.
- Marchini, A.; Ogata, T.; Rappold, G.A. A Track Record on SHOX: From Basic Research to Complex Models and Therapy. Endocr. Rev. 2016, 37, 417–448.
- Ottesen, A.M.; Aksglaede, L.; Garn, I.; Tartaglia, N.; Tassone, F.; Gravholt, C.H.; Bojesen, A.; Sorensen, K.; Jorgensen, N.; Rajpert-De Meyts, E.; et al. Increased number of sex chromosomes affects height in a nonlinear fashion: A study of 305 patients with sex chromosome aneuploidy. Am. J. Med. Genet. A 2010, 152A, 1206–1212.
- Brown, C.J.; Lafreniere, R.G.; Powers, V.E.; Sebastio, G.; Ballabio, A.; Pettigrew, A.L.; Ledbetter, D.H.; Levy, E.; Craig, I.W.; Willard, H.F. Localization of the X inactivation centre on the human X chromosome in Xq13. Nature 1991, 349, 82–84.
- Ahern, D.T.; Bansal, P.; Armillei, M.K.; Faustino, I.V.; Kondaveeti, Y.; Glatt-Deeley, H.R.; Banda, E.C.; Pinter, S.F. Monosomy X in isogenic human iPSC-derived trophoblast model impacts expression modules preserved in human placenta. Proc. Natl. Acad. Sci. USA 2022, 119, e2211073119.
- Zhang, Y.; Castillo-Morales, A.; Jiang, M.; Zhu, Y.; Hu, L.; Urrutia, A.O.; Kong, X.; Hurst, L.D. Genes That Escape X-Inactivation in Humans Have High Intraspecific Variability in Expression, Are Associated with Mental Impairment but Are Not Slow Evolving. Mol. Biol. Evol. 2013, 30, 2588–2601.
- Binder, G. Short stature due to SHOX deficiency: Genotype, phenotype, and therapy. Horm. Res. Paediatr. 2011, 75, 81–89.
- Davies, W. The contribution of Xp22.31 gene dosage to Turner and Klinefelter syndromes and sex-biased phenotypes. Eur. J. Med. Genet. 2021, 64, 104169.
- Rajpathak, S.N.; Vellarikkal, S.K.; Patowary, A.; Scaria, V.; Sivasubbu, S.; Deobagkar, D.D. Human 45,X fibroblast transcriptome reveals distinct differentially expressed genes including long noncoding RNAs potentially associated with the pathophysiology of Turner syndrome. PLoS ONE 2014, 9, e100076.
- Zhang, R.; Hao, L.; Wang, L.; Chen, M.; Li, W.; Li, R.; Yu, J.; Xiao, J.; Wu, J. Gene expression analysis of induced pluripotent stem cells from aneuploid chromosomal syndromes. BMC Genom. 2013, 14, S8.
- Zhang, X.; Hong, D.; Ma, S.; Ward, T.; Ho, M.; Pattni, R.; Duren, Z.; Stankov, A.; Bade Shrestha, S.; Hallmayer, J.; et al. Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc. Natl. Acad. Sci. USA 2020, 117, 4864–4873.
- Wang, H.; Zhu, H.; Zhu, W.; Xu, Y.; Wang, N.; Han, B.; Song, H.; Qiao, J. Bioinformatic Analysis Identifies Potential Key Genes in the Pathogenesis of Turner Syndrome. Front Endocrinol. 2020, 11, 104.
- Urbach, A.; Benvenisty, N. Studying early lethality of 45,XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS ONE 2009, 4, e4175.
- Qi, X.; Wang, Q.; Yu, M.; Kong, Y.; Shi, F.; Wang, S. Bioinformatic analysis identifies the immunological profile of turner syndrome with different X chromosome origins. Front Endocrinol. 2023, 14, 1024244.
- Viuff, M.H.; Berglund, A.; Juul, S.; Andersen, N.H.; Stochholm, K.; Gravholt, C.H. Sex Hormone Replacement Therapy in Turner Syndrome: Impact on Morbidity and Mortality. J. Clin. Endocrinol. Metab. 2020, 105, 468–478.
- Gravholt, C.H.; Juul, S.; Naeraa, R.W.; Hansen, J. Morbidity in Turner syndrome. J. Clin. Epidemiol. 1998, 51, 147–158.
- Schoemaker, M.J.; Swerdlow, A.J.; Higgins, C.D.; Wright, A.F.; Jacobs, P.A.; United Kingdom Clinical Cytogenetics, G. Mortality in women with turner syndrome in Great Britain: A national cohort study. J. Clin. Endocrinol. Metab. 2008, 93, 4735–4742.
- Tuke, M.A.; Ruth, K.S.; Wood, A.R.; Beaumont, R.N.; Tyrrell, J.; Jones, S.E.; Yaghootkar, H.; Turner, C.L.S.; Donohoe, M.E.; Brooke, A.M.; et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet. Med. 2019, 21, 877–886.
- Cameron-Pimblett, A.; La Rosa, C.; King, T.F.J.; Davies, M.C.; Conway, G.S. The Turner syndrome life course project: Karyotype-phenotype analyses across the lifespan. Clin. Endocrinol. 2017, 87, 532–538.
- Bernard, V.; Donadille, B.; Zenaty, D.; Courtillot, C.; Salenave, S.; Brac de la Perriere, A.; Albarel, F.; Fevre, A.; Kerlan, V.; Brue, T.; et al. Spontaneous fertility and pregnancy outcomes amongst 480 women with Turner syndrome. Hum. Reprod. 2016, 31, 782–788.
- Denes, A.M.; Landin-Wilhelmsen, K.; Wettergren, Y.; Bryman, I.; Hanson, C. The proportion of diploid 46,XX cells increases with time in women with Turner syndrome–A 10-year follow-up study. Genet. Test Mol. Biomark. 2015, 19, 82–87.
- El-Mansoury, M.; Barrenas, M.L.; Bryman, I.; Hanson, C.; Larsson, C.; Wilhelmsen, L.; Landin-Wilhelmsen, K. Chromosomal mosaicism mitigates stigmata and cardiovascular risk factors in Turner syndrome. Clin. Endocrinol. 2007, 66, 744–751.
- Bryman, I.; Sylven, L.; Berntorp, K.; Innala, E.; Bergstrom, I.; Hanson, C.; Oxholm, M.; Landin-Wilhelmsen, K. Pregnancy rate and outcome in Swedish women with Turner syndrome. Fertil. Steril. 2011, 95, 2507–2510.
- Sybert, V.P. Phenotypic effects of mosaicism for a 47,XXX cell line in Turner syndrome. J. Med. Genet. 2002, 39, 217–220.
- Snyder, E.A.; San Roman, A.K.; Pina-Aguilar, R.E.; Steeves, M.A.; McNamara, E.A.; Souter, I.; Hayes, F.J.; Levitsky, L.L.; Lin, A.E. Genetic counseling for women with 45,X/46,XX mosaicism: Towards more personalized management. Eur. J. Med. Genet. 2021, 64, 104140.
- Luo, Y.; Zhu, D.; Du, R.; Gong, Y.; Xie, C.; Xu, X.; Fan, Y.; Yu, B.; Sun, X.; Chen, Y. Uniparental disomy of the entire X chromosome in Turner syndrome patient-specific induced pluripotent stem cells. Cell Discov. 2015, 1, 15022.
- Hovatta, O. Pregnancies in women with Turner’s syndrome. Ann. Med. 1999, 31, 106–110.
- Birkebaek, N.H.; Cruger, D.; Hansen, J.; Nielsen, J.; Bruun-Petersen, G. Fertility and pregnancy outcome in Danish women with Turner syndrome. Clin. Genet. 2002, 61, 35–39.
- Hadnott, T.N.; Gould, H.N.; Gharib, A.M.; Bondy, C.A. Outcomes of spontaneous and assisted pregnancies in Turner syndrome: The U.S. National Institutes of Health experience. Fertil. Steril. 2011, 95, 2251–2256.
- Lippe, B. Turner syndrome. Endocrinol. Metab. Clin. N. Am. 1991, 20, 121–152.
- Cleemann, L.; Holm, K.; Fallentin, E.; Skouby, S.O.; Smedegaard, H.; Moller, N.; Borch-Christensen, H.; Jeppesen, E.M.; Wieslander, S.B.; Andersson, A.M.; et al. Uterus and ovaries in girls and young women with Turner syndrome evaluated by ultrasound and magnetic resonance imaging. Clin. Endocrinol. 2011, 74, 756–761.
- Weiss, L. Additional evidence of gradual loss of germ cells in the pathogenesis of streak ovaries in Turner’s syndrome. J. Med. Genet. 1971, 8, 540–544.
- Prakash, S.K.; Crenshaw, M.L.; Backeljauw, P.F.; Silberbach, M.; Scurlock, C.; Culin, D.D.; Ranallo, K.C.; Lin, A.E. 45,X mosaicism in a population-based biobank: Implications for Turner syndrome. Genet. Med. 2019, 21, 1882–1883.
- Negreiros, L.P.; Bolina, E.R.; Guimaraes, M.M. Pubertal development profile in patients with Turner syndrome. J. Pediatr. Endocrinol. Metab. 2014, 27, 845–849.
- Viuff, M.; Gravholt, C.H. Turner Syndrome and Fertility. Ann. Endocrinol. 2022, 83, 244–249.
- Reynaud, K.; Cortvrindt, R.; Verlinde, F.; De Schepper, J.; Bourgain, C.; Smitz, J. Number of ovarian follicles in human fetuses with the 45,X karyotype. Fertil. Steril. 2004, 81, 1112–1119.
- Hovatta, O. Ovarian function and in vitro fertilization (IVF) in Turner syndrome. Pediatr. Endocrinol. Rev. 2012, 9, 713–717.
- Bakalov, V.K.; Shawker, T.; Ceniceros, I.; Bondy, C.A. Uterine development in Turner syndrome. J. Pediatr. 2007, 151, 528–531.
- Viuff, M.H.; Just, J.; Brun, S.; Dam, T.V.; Hansen, M.; Melgaard, L.; Hougaard, D.M.; Lappe, M.; Gravholt, C.H. Women with Turner Syndrome Are Both Estrogen and Androgen Deficient: The Impact of Hormone Replacement Therapy. J. Clin. Endocrinol. Metab. 2022, 107, 1983–1993.
- Hagen, C.P.; Main, K.M.; Kjaergaard, S.; Juul, A. FSH, LH, inhibin B and estradiol levels in Turner syndrome depend on age and karyotype: Longitudinal study of 70 Turner girls with or without spontaneous puberty. Hum. Reprod. 2010, 25, 3134–3141.
- Ljubicic, M.L.; Busch, A.S.; Upners, E.N.; Fischer, M.B.; Petersen, J.H.; Raket, L.L.; Frederiksen, H.; Johannsen, T.H.; Juul, A.; Hagen, C.P. A Biphasic Pattern of Reproductive Hormones in Healthy Female Infants: The COPENHAGEN Minipuberty Study. J. Clin. Endocrinol. Metab. 2022, 107, 2598–2605.
- Paterson, W.F.; Hollman, A.S.; Donaldson, M.D. Poor uterine development in Turner syndrome with oral oestrogen therapy. Clin. Endocrinol. 2002, 56, 359–365.
- Practice Committee of American Society for Reproductive Medicine. Increased maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome. Fertil. Steril. 2012, 97, 282–284.
- Cauldwell, M.; Steer, P.J.; Adamson, D.; Alexander, C.; Allen, L.; Bhagra, C.; Bolger, A.; Bonner, S.; Calanchini, M.; Carroll, A.; et al. Pregnancies in women with Turner syndrome: A retrospective multicentre UK study. BJOG 2022, 129, 796–803.
- Silberbach, M.; Roos-Hesselink, J.W.; Andersen, N.H.; Braverman, A.C.; Brown, N.; Collins, R.T.; De Backer, J.; Eagle, K.A.; Hiratzka, L.F.; Johnson, W.H., Jr.; et al. Cardiovascular Health in Turner Syndrome: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000048.
- Miyabara, S.; Nakayama, M.; Suzumori, K.; Yonemitsu, N.; Sugihara, H. Developmental analysis of cardiovascular system of 45,X fetuses with cystic hygroma. Am. J. Med. Genet. 1997, 68, 135–141.
- Angelini, P. Coronary artery anomalies: An entity in search of an identity. Circulation 2007, 115, 1296–1305.
- Koenraadt, W.M.C.; Siebelink, H.J.; Bartelings, M.M.; Schalij, M.J.; van der Vlugt, M.J.; van den Bosch, A.E.; Budde, R.P.J.; Roos-Hesselink, J.W.; Duijnhouwer, A.L.; van den Hoven, A.T.; et al. Coronary anatomy in Turner syndrome versus patients with isolated bicuspid aortic valves. Heart 2019, 105, 701–707.
- Viuff, M.H.; Trolle, C.; Wen, J.; Jensen, J.M.; Norgaard, B.L.; Gutmark, E.J.; Gutmark-Little, I.; Mortensen, K.H.; Gravholt, C.H.; Andersen, N.H. Coronary artery anomalies in Turner Syndrome. J. Cardiovasc. Comput. Tomogr. 2016, 10, 480–484.
- Zakaria, D.; Tang, X.; Bhakta, R.; ElHassan, N.O.; Prodhan, P. Chromosomal Abnormalities Affect the Surgical Outcome in Infants with Hypoplastic Left Heart Syndrome: A Large Cohort Analysis. Pediatr. Cardiol. 2018, 39, 11–18.
- Phillips, H.M.; Mahendran, P.; Singh, E.; Anderson, R.H.; Chaudhry, B.; Henderson, D.J. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 2013, 99, 452–460.
- Van Nisselrooij, A.E.L.; Lugthart, M.A.; Clur, S.A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Rozendaal, L.; Blom, N.A.; van Lith, J.M.M.; Knegt, A.C.; et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet. Med. 2020, 22, 1206–1214.
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth Muscle Cells Derived from Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726.
- Mortensen, K.H.; Hjerrild, B.E.; Andersen, N.H.; Sorensen, K.E.; Horlyck, A.; Pedersen, E.M.; Lundorf, E.; Christiansen, J.S.; Gravholt, C.H. Abnormalities of the major intrathoracic arteries in Turner syndrome as revealed by magnetic resonance imaging. Cardiol. Young 2010, 20, 191–200.
- Kim, H.K.; Gottliebson, W.; Hor, K.; Backeljauw, P.; Gutmark-Little, I.; Salisbury, S.R.; Racadio, J.M.; Helton-Skally, K.; Fleck, R. Cardiovascular anomalies in Turner syndrome: Spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. AJR Am. J. Roentgenol. 2011, 196, 454–460.
- Ghazi Sherbaf, F.; Mohajer, B.; Ashraf-Ganjouei, A.; Mojtahed Zadeh, M.; Javinani, A.; Sanjari Moghaddam, H.; Shirin Shandiz, M.; Aarabi, M.H. Serum Insulin-Like Growth Factor-1 in Parkinson’s Disease; Study of Cerebrospinal Fluid Biomarkers and White Matter Microstructure. Front. Endocrinol. 2018, 9, 608.
- Kruger, T.; Forkavets, O.; Veseli, K.; Lausberg, H.; Vohringer, L.; Schneider, W.; Bamberg, F.; Schlensak, C. Ascending aortic elongation and the risk of dissection. Eur. J. Cardiothorac. Surg. 2016, 50, 241–247.
- Patel, A.; Costello, J.M.; Backer, C.L.; Pasquali, S.K.; Hill, K.D.; Wallace, A.S.; Jacobs, J.P.; Jacobs, M.L. Prevalence of Noncardiac and Genetic Abnormalities in Neonates Undergoing Cardiac Operations: Analysis of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2016, 102, 1607–1614.
- Mortensen, K.H.; Hjerrild, B.E.; Stochholm, K.; Andersen, N.H.; Sorensen, K.E.; Lundorf, E.; Horlyck, A.; Pedersen, E.M.; Christiansen, J.S.; Gravholt, C.H. Dilation of the ascending aorta in Turner syndrome–a prospective cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2011, 13, 24.
- Ostberg, J.E.; Donald, A.E.; Halcox, J.P.; Storry, C.; McCarthy, C.; Conway, G.S. Vasculopathy in Turner syndrome: Arterial dilatation and intimal thickening without endothelial dysfunction. J. Clin. Endocrinol. Metab. 2005, 90, 5161–5166.
- Dulac, Y.; Pienkowski, C.; Abadir, S.; Tauber, M.; Acar, P. Cardiovascular abnormalities in Turner’s syndrome: What prevention? Arch. Cardiovasc. Dis. 2008, 101, 485–490.
- Roulot, D.; Degott, C.; Chazouilleres, O.; Oberti, F.; Cales, P.; Carbonell, N.; Benferhat, S.; Bresson-Hadni, S.; Valla, D. Vascular involvement of the liver in Turner’s syndrome. Hepatology 2004, 39, 239–247.
- Corbitt, H.; Morris, S.A.; Gravholt, C.H.; Mortensen, K.H.; Tippner-Hedges, R.; Silberbach, M.; Maslen, C.L.; Gen, T.A.C.R.I. TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genet. 2018, 14, e1007692.
- Rabkin, S.W. Differential expression of MMP-2, MMP-9 and TIMP proteins in thoracic aortic aneurysm–comparison with and without bicuspid aortic valve: A meta-analysis. Vasa 2014, 43, 433–442.
- Trolle, C.; Nielsen, M.M.; Skakkebaek, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Orntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220.
- Albareda, M.M.; Gallego, A.; Enriquez, J.; Rodriguez, J.L.; Webb, S.M. Biochemical liver abnormalities in Turner’s syndrome. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1037–1039.
- Floreani, A.; Molaro, M.; Baragiotta, A.; Naccarato, R. Chronic cholestasis associated with Turner’s syndrome. Digestion 1999, 60, 587–589.
- Salerno, M.; Di Maio, S.; Gasparini, N.; Rizzo, M.; Ferri, P.; Vajro, P. Liver abnormalities in Turner syndrome. Eur. J. Pediatr. 1999, 158, 618–623.
- Gardner, L.I. Letter: Intrahepatic bile stasis in 45,X Turner’s syndrome. N. Engl. J. Med. 1974, 290, 406.
- Krivosheev, A.B. Development of liver cirrhosis in a female patient with Shereshevskii-Turner syndrome. Klin. Med. 1990, 68, 95–96.
- Garavelli, L.; Donadio, A.; Banchini, G.; Fornaciari, G.; Plancher, A.C.; Franchi, F.; Gardini, G. Liver abnormalities and portal hypertension in Ullrich-Turner syndrome. Am. J. Med. Genet. 1998, 80, 180–182.
- Singh, I.; Noel, G.; Barker, J.M.; Chatfield, K.C.; Furniss, A.; Khanna, A.D.; Nokoff, N.J.; Patel, S.; Pyle, L.; Nahata, L.; et al. Hepatic abnormalities in youth with Turner syndrome. Liver Int. 2022, 42, 2237–2246.
- Machlab, S.; Miquel, M.; Volta, T.; Escoda, M.R.; Vergara, M. Turner syndrome as a cause of liver cirrhosis. Gastroenterol. Hepatol. 2018, 41, 308–309.
- El-Mansoury, M.; Berntorp, K.; Bryman, I.; Hanson, C.; Innala, E.; Karlsson, A.; Landin-Wilhelmsen, K. Elevated liver enzymes in Turner syndrome during a 5-year follow-up study. Clin. Endocrinol. 2008, 68, 485–490.
- Larizza, D.; Locatelli, M.; Vitali, L.; Vigano, C.; Calcaterra, V.; Tinelli, C.; Sommaruga, M.G.; Bozzini, A.; Campani, R.; Severi, F. Serum liver enzymes in Turner syndrome. Eur. J. Pediatr. 2000, 159, 143–148.
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Engl. J. Med. 2004, 351, 1227–1238.
- Izumita, Y.; Nishigaki, S.; Satoh, M.; Takubo, N.; Numakura, C.; Takahashi, I.; Soneda, S.; Abe, Y.; Kamasaki, H.; Ohtsu, Y.; et al. Retrospective study of the renal function using estimated glomerular filtration rate and congenital anomalies of the kidney-urinary tract in pediatric Turner syndrome. Congenit. Anom. 2020, 60, 175–179.
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70.
- Ogawa, T.; Takizawa, F.; Mukoyama, Y.; Ogawa, A.; Ito, J. Renal morphology and function from childhood to adulthood in Turner syndrome. Clin. Exp. Nephrol. 2021, 25, 633–640.
- Egli, F.; Stalder, G. Malformations of kidney and urinary tract in common chromosomal aberrations. I. Clinical studies. Humangenetik 1973, 18, 1–15.
- Gregoir, W. Conservative Surgery in Horseshoe-Kidney. Urol. Int. 1963, 16, 129–138.
- Pritti, K.; Mishra, V.; Patel, H. A Rare Case of Mosaic Ring Turner Syndrome with Horseshoe Kidney. J. Hum. Reprod. Sci. 2022, 15, 318–320.
- Bilge, I.; Kayserili, H.; Emre, S.; Nayir, A.; Sirin, A.; Tukel, T.; Bas, F.; Kilic, G.; Basaran, S.; Gunoz, H.; et al. Frequency of renal malformations in Turner syndrome: Analysis of 82 Turkish children. Pediatr. Nephrol. 2000, 14, 1111–1114.
- Horita, S.; Simsek, E.; Simsek, T.; Yildirim, N.; Ishiura, H.; Nakamura, M.; Satoh, N.; Suzuki, A.; Tsukada, H.; Mizuno, T.; et al. SLC4A4 compound heterozygous mutations in exon-intron boundary regions presenting with severe proximal renal tubular acidosis and extrarenal symptoms coexisting with Turner’s syndrome: A case report. BMC Med. Genet. 2018, 19, 103.
- Cintron, D.; Rodriguez-Gutierrez, R.; Serrano, V.; Latortue-Albino, P.; Erwin, P.J.; Murad, M.H. Effect of estrogen replacement therapy on bone and cardiovascular outcomes in women with turner syndrome: A systematic review and meta-analysis. Endocrine 2017, 55, 366–375.
- Gravholt, C.H. Aspects of the treatment of Turner syndrome. Expert Opin. Pharmacother. 2001, 2, 1633–1647.
- Gussinye, M.; Terrades, P.; Yeste, D.; Vicens-Calvet, E.; Carrascosa, A. Low areal bone mineral density values in adolescents and young adult turner syndrome patients increase after long-term transdermal estradiol therapy. Horm. Res. 2000, 54, 131–135.
- Elsheikh, M.; Conway, G.S.; Wass, J.A. Medical problems in adult women with Turner’s syndrome. Ann. Med. 1999, 31, 99–105.
- Saenger, P.; Wikland, K.A.; Conway, G.S.; Davenport, M.; Gravholt, C.H.; Hintz, R.; Hovatta, O.; Hultcrantz, M.; Landin-Wilhelmsen, K.; Lin, A.; et al. Recommendations for the diagnosis and management of Turner syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3061–3069.
- Landin-Wilhelmsen, K.; Bryman, I.; Windh, M.; Wilhelmsen, L. Osteoporosis and fractures in Turner syndrome-importance of growth promoting and oestrogen therapy. Clin. Endocrinol. 1999, 51, 497–502.
- Even, L.; Bronstein, V.; Hochberg, Z. Bone maturation in girls with Turner’s syndrome. Eur. J. Endocrinol. 1998, 138, 59–62.
- Holroyd, C.R.; Davies, J.H.; Taylor, P.; Jameson, K.; Rivett, C.; Cooper, C.; Dennison, E.M. Reduced cortical bone density with normal trabecular bone density in girls with Turner syndrome. Osteoporos Int. 2010, 21, 2093–2099.
- Wasserman, H.; Backeljauw, P.F.; Khoury, J.C.; Kalkwarf, H.J.; Gordon, C.M. Bone fragility in Turner syndrome: Fracture prevalence and risk factors determined by a national patient survey. Clin. Endocrinol. 2018, 89, 46–55.
- Bakalov, V.K.; Chen, M.L.; Baron, J.; Hanton, L.B.; Reynolds, J.C.; Stratakis, C.A.; Axelrod, L.E.; Bondy, C.A. Bone mineral density and fractures in Turner syndrome. Am. J. Med. 2003, 115, 259–264.
- Soucek, O.; Lebl, J.; Snajderova, M.; Kolouskova, S.; Rocek, M.; Hlavka, Z.; Cinek, O.; Rittweger, J.; Sumnik, Z. Bone geometry and volumetric bone mineral density in girls with Turner syndrome of different pubertal stages. Clin. Endocrinol. 2011, 74, 445–452.
- Bakalov, V.K.; Axelrod, L.; Baron, J.; Hanton, L.; Nelson, L.M.; Reynolds, J.C.; Hill, S.; Troendle, J.; Bondy, C.A. Selective reduction in cortical bone mineral density in turner syndrome independent of ovarian hormone deficiency. J. Clin. Endocrinol. Metab. 2003, 88, 5717–5722.
- Reiss, A.L.; Mazzocco, M.M.; Greenlaw, R.; Freund, L.S.; Ross, J.L. Neurodevelopmental effects of X monosomy: A volumetric imaging study. Ann. Neurol. 1995, 38, 731–738.
- Murphy, D.G.; DeCarli, C.; Daly, E.; Haxby, J.V.; Allen, G.; White, B.J.; McIntosh, A.R.; Powell, C.M.; Horwitz, B.; Rapoport, S.I.; et al. X-chromosome effects on female brain: A magnetic resonance imaging study of Turner’s syndrome. Lancet 1993, 342, 1197–1200.
- Reiss, A.L.; Freund, L.; Plotnick, L.; Baumgardner, T.; Green, K.; Sozer, A.C.; Reader, M.; Boehm, C.; Denckla, M.B. The effects of X monosomy on brain development: Monozygotic twins discordant for Turner’s syndrome. Ann. Neurol. 1993, 34, 95–107.
- Kesler, S.R.; Blasey, C.M.; Brown, W.E.; Yankowitz, J.; Zeng, S.M.; Bender, B.G.; Reiss, A.L. Effects of X-monosomy and X-linked imprinting on superior temporal gyrus morphology in Turner syndrome. Biol. Psychiatry 2003, 54, 636–646.
- Lepage, J.F.; Mazaika, P.K.; Hong, D.S.; Raman, M.; Reiss, A.L. Cortical brain morphology in young, estrogen-naive, and adolescent, estrogen-treated girls with Turner syndrome. Cereb. Cortex 2013, 23, 2159–2168.
- Marzelli, M.J.; Hoeft, F.; Hong, D.S.; Reiss, A.L. Neuroanatomical spatial patterns in Turner syndrome. Neuroimage 2011, 55, 439–447.
- Knickmeyer, R.C. Turner syndrome: Advances in understanding altered cognition, brain structure and function. Curr. Opin. Neurol. 2012, 25, 144–149.
- Heard, E.; Chaumeil, J.; Masui, O.; Okamoto, I. Mammalian X-chromosome inactivation: An epigenetics paradigm. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 89–102.
- Sahakyan, A.; Kim, R.; Chronis, C.; Sabri, S.; Bonora, G.; Theunissen, T.W.; Kuoy, E.; Langerman, J.; Clark, A.T.; Jaenisch, R.; et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem. Cell 2017, 20, 87–101.
- Berletch, J.B.; Yang, F.; Disteche, C.M. Escape from X inactivation in mice and humans. Genome. Biol. 2010, 11, 213.
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes that escape from X inactivation. Hum. Genet. 2011, 130, 237–245.
- Backeljauw, P.; Chernausek, S.D.; Gravholt, C.H.; Kruszka, P. Turner syndrome. In Sperling Pediatric Endocrinology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 627–660.
- Jones, M.H.; Furlong, R.A.; Burkin, H.; Chalmers, I.J.; Brown, G.M.; Khwaja, O.; Affara, N.A. The Drosophila developmental gene fat facets has a human homologue in Xp11.4 which escapes X-inactivation and has related sequences on Yq11.2. Hum. Mol. Genet. 1996, 5, 1695–1701.
- Quilter, C.R.; Karcanias, A.C.; Bagga, M.R.; Duncan, S.; Murray, A.; Conway, G.S.; Sargent, C.A.; Affara, N.A. Analysis of X chromosome genomic DNA sequence copy number variation associated with premature ovarian failure (POF). Hum. Reprod. 2010, 25, 2139–2150.
- Laupacis, A.; Bourne, R.; Rorabeck, C.; Feeny, D.; Tugwell, P.; Wong, C. Comparison of total hip arthroplasty performed with and without cement: A randomized trial. J. Bone Joint Surg. Am. 2002, 84, 1823–1828.
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013, 34, 108–110.
- Alvarez-Nava, F.; Lanes, R. Epigenetics in Turner syndrome. Clin. Epigenetics 2018, 10, 45.
More
Information
Subjects:
Developmental Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
2 times
(View History)
Update Date:
23 May 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No