Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jeong Tae Do and Version 2 by Catherine Yang.
Turner syndrome (TS), a genetic disorder due to incomplete dosage compensation of X-linked genes, affects multiple organ systems, leading to hypogonadotropic hypogonadism, short stature, cardiovascular and vascular abnormalities, liver disease, renal abnormalities, brain abnormalities, and skeletal problems. Patients with TS experience premature ovarian failure with a rapid decline in ovarian function caused by germ cell depletion, and pregnancies carry a high risk of adverse maternal and fetal outcomes. Aortic abnormalities, heart defects, obesity, hypertension, and liver abnormalities, such as steatosis, steatohepatitis, biliary involvement, liver cirrhosis, and nodular regenerative hyperplasia, are commonly observed in patients with TS.
- Turner syndrome
- X monosomy
- X chromosome inactivation
- organ abnormalities
1. Introduction
Turner syndrome (TS) is one of the most common disorders caused by chromosomal abnormalities, affecting approximately 1 in 2500 live female births. It is the only viable monosomy syndrome caused by partial or complete loss of one of the two sex chromosomes [1]. TS was first reported in 1938 by Henry H. Turner as a syndrome of infantilism, congenital webbed neck, and cubitus valgus, and Ford et al. found that the disease was caused by sex chromosomal abnormality in 1959 [2][3][2,3]. The most common karyotype in TS is 45,X, accounting for 40–50% of all cases of TS, whereas 45,X/46,XX or 45,X/47,XXX mosaicism account for 20–30%. The remaining cases include Y chromosome variants and X chromosome structural abnormalities, such as isochromosome Xq, deletion of Xp or Xq (which can occur as mosaicism), and ring X (which is always mosaic) [4] (Figure 1). Thus, in TS, only one X chromosome is normal and the others are absent or abnormal. The diagnosis of TS has traditionally relied on the clinical phenotype in addition to standard chromosomal analysis [5]. Total or partial loss of one of the two sex chromosomes affects biological pathways and networks [5], and, in some cases, SHOX gene defects have been linked to certain phenotypes of TS [6][7][6,7] (Table 1).
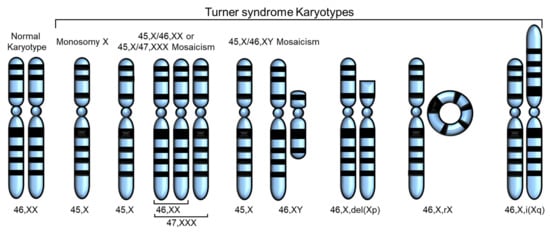
Figure 1. Various karyotypes of Turner syndrome (TS) modified from Huang et al. [8]. Partial or complete loss of the second sex chromosome results in TS. The most common karyotype in TS is monosomy X (45,X), and the others are 45,X/46,XX or 45,X/47,XXX mosaicism, 45,X/45,XY mosaicism, deletion of Xp or Xq, ring X (46,X,rX), and isochromosome Xq.
Table 1.
Genes associated with Turner Syndrome (TS).
| Gene | Location | Expression in TS | Associated Phenotype | Reference |
|---|---|---|---|---|
| SHOX | Xp22.33 and Y chromosome (PAR1) | Decreased expression | Short stature, Madelung wrist deformity, Intellectual disabilities |
[9][10][11][12][13][9,10,11,12,13] |
| ARSD, ARSE, ARSF | Xp22.3 | Loss owing to contiguous gene deletion syndrome | Chondrodysplasia punctata | [14][15][16][14,15,16] |
| STS | Xp22.31 | Loss owing to contiguous gene deletion syndrome | X-linked ichthyosis | [13][14][15][16][13,14,15,16] |
| GPR143 | Xp22.2 | Loss owing to contiguous gene deletion syndrome | Ocular albinism type I | [14][15][16][14,15,16] |
| ANOS1 | Xp23.3 | Loss owing to contiguous gene deletion syndrome | Kallmann syndrome | [14][15][16][14,15,16] |
| RPS4X | Xq13.1 | Downregulation | N/A | [13][17][18][13[,1719,18],19] |
| CD99 | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20][13,20] |
| CSF2RA | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20][21][22][13,20,21,22] |
| MYL9 | 20q11.23 | Downregulated | N/A | [20] |
| MYLPF | 16p11.2 | Downregulated | N/A | [20] |
| IGFBP2 | 2q35 | Downregulated | N/A | [20] |
Individuals with TS are at an increased risk of endocrine diagnoses, including diabetes, thyroid and parathyroid disorders, celiac disease, and osteoporosis [23][24][23,24], as well as cardiovascular diseases, including arrhythmia, ischemic heart disease, hypertension, hyperlipidemia, and stroke. This is supported by the increased use of prescription drugs by patients with TS [25]. The 45,X karyotype is associated with the highest rates of morbidity and mortality, whereas the mosaic karyotype is associated with a low prevalence for cardiovascular, metabolic, renal, and reproductive phenotypes [26][27][28][29][30][31][32][33][26,27,28,29,30,31,32,33]. Despite ongoing research, no feasible treatment has been proposed owing to the severe effects of losing an entire chromosome and the numerous genes that are simultaneously affected [34].
2. Fertility Problems
Infertility is one of the most common symptoms of TS, despite low rates of spontaneous pregnancies [31][35][36][37][31,35,36,37]. TS is accompanied by hypogonadotropic hypogonadism in almost all patients, leading to primary or secondary amenorrhea and infertility owing to premature ovarian failure (POF) (affecting approximately 95% of women with TS) and premature ovarian insufficiency [28][38][39][28,38,39]. Therefore, women with TS do not produce enough eggs or the necessary hormones to support pregnancy. The ovaries in a 45,X fetus appear to develop normally until birth; however, follicular atresia is induced by birth or early childhood [40]. Moreover, 5–20% of girls with TS retain enough follicles to permit spontaneous menarche, even if early menopause typically follows. Women with TS who have a mosaic karyotype, or experience spontaneous puberty, have follicles in one or both ovaries [39]. Furthermore, those with low levels of 45,X/46,XX mosaicism are less severely affected and have a high likelihood of experiencing spontaneous menstruation and pregnancy, although karyotype does not always predict phenotype [41][42][43][41,42,43]. Accelerated germ cell death is presumed to be the major mechanism causing germ cell depletion in patients with TS.
Reynaud et al. analyzed 10 aborted fetuses with TS and found that the number of germ cells in the genital ridge was similar to that in the control group up to 12 weeks of gestation, indicating normal migration of primordial germ cells in fetuses with TS [44]. However, differences were observed from 18 weeks of gestation, where germ cells were rarely detected, and completely absent at 25 weeks of gestation in fetuses with 45,X TS. Moreover, primordial and antral follicles were absent in fetuses with 45,X TS, although they were present in fetuses with TS with mosaicism. These studies suggest that folliculogenesis is severely impaired in ovaries of patients with TS, possibly owing to the loss of germ cells [44]. Additionally, the eggs from women with TS might be of poor quality, decreasing the chances of successful fertilization and pregnancy [45].
TS can also cause abnormalities in the structure and function of the uterus, affecting the implantation and growth of fertilized eggs [46]. Only about a quarter of people with TS have a fully developed uterus in size and shape, while most others have a slightly smaller uterus; about one-third have an immature form of the uterus. Notably, the difference in the size of the uterus between women with TS and those with a normal karyotype is not significant; however, on average, women with TS have a smaller uterine volume than those with a normal karyotype. The size of the uterus in individuals with TS can be influenced by various factors, including the age of the patient, duration of estrogen use, use of hormone replacement therapy (HRT), and type of estrogen medication administered. However, with appropriate and timely treatment, women with TS can achieve normal uterine development [46].
In addition, an imbalance in sex hormone levels affects the fertility of patients with TS. Women with TS showed 30–50% lower levels of androgens, including testosterone, free androgen index, androstenedione, and dehydroepiandrosterone sulfate, than those with a normal karyotype, but an increase in Follicle stimulating hormone (FSH), Luteinizing hormone (LH), and estrone sulfate levels up to twice the normal range [47]. High levels of FSH and LH during adolescence are linked to reduced ovarian function [48]. However, patients with TS showed a normal biphasic age pattern of reproductive hormones, with peak FSH and LH levels occurring at three months of age, followed by a subsequent decrease to minimal levels during mid-childhood and reactivation at puberty [48][49][48,49].
Pregnancy is rare among patients with TS and shows a high risk of miscarriage, stillbirth, and birth defects [50]. Only 2–5% of patients with TS become pregnant spontaneously, and approximately 3.8% of patients with TS have one or more live-born children [28][35][28,35]. Both natural and medically assisted pregnancies in patients with TS have a higher risk of adverse maternal and fetal outcomes than those in healthy women. For instance, 23–50% of women with TS have congenital heart disease, and pregnancy causes a 50% increase in cardiac output, making patients with TS susceptible to aortic dissection or rupture. As a result, the risk of death during pregnancy for patients with TS can reach up to 2% [51][52][51,52].