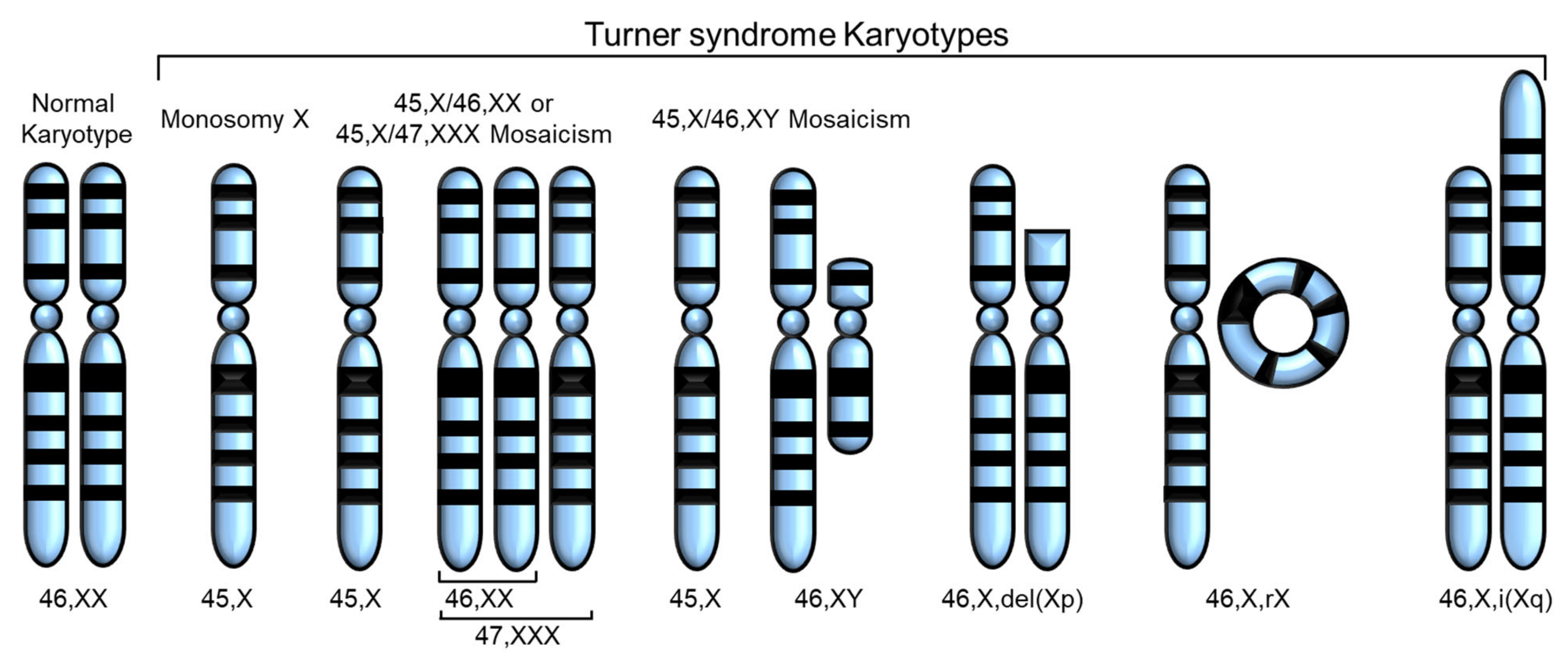
Turner syndrome (TS), a genetic disorder due to incomplete dosage compensation of X-linked genes, affects multiple organ systems, leading to hypogonadotropic hypogonadism, short stature, cardiovascular and vascular abnormalities, liver disease, renal abnormalities, brain abnormalities, and skeletal problems. Patients with TS experience premature ovarian failure with a rapid decline in ovarian function caused by germ cell depletion, and pregnancies carry a high risk of adverse maternal and fetal outcomes. Aortic abnormalities, heart defects, obesity, hypertension, and liver abnormalities, such as steatosis, steatohepatitis, biliary involvement, liver cirrhosis, and nodular regenerative hyperplasia, are commonly observed in patients with TS.
- Turner syndrome
- X monosomy
- X chromosome inactivation
- organ abnormalities
1. Introduction
| Gene | Location | Expression in TS | Associated Phenotype | Reference |
|---|---|---|---|---|
| SHOX | Xp22.33 and Y chromosome (PAR1) | Decreased expression | Short stature, Madelung wrist deformity, Intellectual disabilities |
[9][10][11][12][13] |
| ARSD, ARSE, ARSF | Xp22.3 | Loss owing to contiguous gene deletion syndrome | Chondrodysplasia punctata | [14][15][16] |
| STS | Xp22.31 | Loss owing to contiguous gene deletion syndrome | X-linked ichthyosis | [13][14][15][16] |
| GPR143 | Xp22.2 | Loss owing to contiguous gene deletion syndrome | Ocular albinism type I | [14][15][16] |
| ANOS1 | Xp23.3 | Loss owing to contiguous gene deletion syndrome | Kallmann syndrome | [14][15][16] |
| RPS4X | Xq13.1 | Downregulation | N/A | [13][17][18][19] |
| CD99 | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20] |
| CSF2RA | X and Y chromosomes (PAR1) | Downregulation | N/A | [13][20][21][22] |
| MYL9 | 20q11.23 | Downregulated | N/A | [20] |
| MYLPF | 16p11.2 | Downregulated | N/A | [20] |
| IGFBP2 | 2q35 | Downregulated | N/A | [20] |
2. Fertility Problems
3. Heart and Cardiovascular Disease
4. Liver Abnormalities
5. Kidney Abnormalities
6. Skeletal Abnormalities and Short Stature
7. Brain Abnormalities
8. Relevance to X Chromosome Inactivation and Escape Genes
This entry is adapted from the peer-reviewed paper 10.3390/cells12101365
References
- Saenger, P. Turner’s syndrome. N. Engl. J. Med. 1996, 335, 1749–1754.
- Turner, H.H. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938, 23, 566–574.
- Ford, C.E.; Jones, K.W.; Polani, P.E.; De Almeida, J.C.; Briggs, J.H. A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner’s syndrome). Lancet 1959, 1, 711–713.
- Bollig, K.J.; Mainigi, M.; Senapati, S.; Lin, A.E.; Levitsky, L.L.; Bamba, V. Turner syndrome: Fertility counselling in childhood and through the reproductive lifespan. Curr. Opin. Endocrinol. Diabetes Obes. 2023, 30, 16–26.
- Gravholt, C.H.; Viuff, M.; Just, J.; Sandahl, K.; Brun, S.; van der Velden, J.; Andersen, N.H.; Skakkebaek, A. The Changing Face of Turner Syndrome. Endocr. Rev. 2023, 44, 33–69.
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 1997, 16, 54–63.
- Ellison, J.W.; Wardak, Z.; Young, M.F.; Gehron Robey, P.; Laig-Webster, M.; Chiong, W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum. Mol. Genet. 1997, 6, 1341–1347.
- Huang, A.C.; Olson, S.B.; Maslen, C.L. A Review of Recent Developments in Turner Syndrome Research. J. Cardiovasc. Dev. Dis. 2021, 8, 138.
- Binder, G.; Fritsch, H.; Schweizer, R.; Ranke, M.B. Radiological signs of Leri-Weill dyschondrosteosis in Turner syndrome. Horm. Res. 2001, 55, 71–76.
- Marchini, A.; Ogata, T.; Rappold, G.A. A Track Record on SHOX: From Basic Research to Complex Models and Therapy. Endocr. Rev. 2016, 37, 417–448.
- Ottesen, A.M.; Aksglaede, L.; Garn, I.; Tartaglia, N.; Tassone, F.; Gravholt, C.H.; Bojesen, A.; Sorensen, K.; Jorgensen, N.; Rajpert-De Meyts, E.; et al. Increased number of sex chromosomes affects height in a nonlinear fashion: A study of 305 patients with sex chromosome aneuploidy. Am. J. Med. Genet. A 2010, 152A, 1206–1212.
- Brown, C.J.; Lafreniere, R.G.; Powers, V.E.; Sebastio, G.; Ballabio, A.; Pettigrew, A.L.; Ledbetter, D.H.; Levy, E.; Craig, I.W.; Willard, H.F. Localization of the X inactivation centre on the human X chromosome in Xq13. Nature 1991, 349, 82–84.
- Ahern, D.T.; Bansal, P.; Armillei, M.K.; Faustino, I.V.; Kondaveeti, Y.; Glatt-Deeley, H.R.; Banda, E.C.; Pinter, S.F. Monosomy X in isogenic human iPSC-derived trophoblast model impacts expression modules preserved in human placenta. Proc. Natl. Acad. Sci. USA 2022, 119, e2211073119.
- Zhang, Y.; Castillo-Morales, A.; Jiang, M.; Zhu, Y.; Hu, L.; Urrutia, A.O.; Kong, X.; Hurst, L.D. Genes That Escape X-Inactivation in Humans Have High Intraspecific Variability in Expression, Are Associated with Mental Impairment but Are Not Slow Evolving. Mol. Biol. Evol. 2013, 30, 2588–2601.
- Binder, G. Short stature due to SHOX deficiency: Genotype, phenotype, and therapy. Horm. Res. Paediatr. 2011, 75, 81–89.
- Davies, W. The contribution of Xp22.31 gene dosage to Turner and Klinefelter syndromes and sex-biased phenotypes. Eur. J. Med. Genet. 2021, 64, 104169.
- Rajpathak, S.N.; Vellarikkal, S.K.; Patowary, A.; Scaria, V.; Sivasubbu, S.; Deobagkar, D.D. Human 45,X fibroblast transcriptome reveals distinct differentially expressed genes including long noncoding RNAs potentially associated with the pathophysiology of Turner syndrome. PLoS ONE 2014, 9, e100076.
- Zhang, R.; Hao, L.; Wang, L.; Chen, M.; Li, W.; Li, R.; Yu, J.; Xiao, J.; Wu, J. Gene expression analysis of induced pluripotent stem cells from aneuploid chromosomal syndromes. BMC Genom. 2013, 14, S8.
- Zhang, X.; Hong, D.; Ma, S.; Ward, T.; Ho, M.; Pattni, R.; Duren, Z.; Stankov, A.; Bade Shrestha, S.; Hallmayer, J.; et al. Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc. Natl. Acad. Sci. USA 2020, 117, 4864–4873.
- Wang, H.; Zhu, H.; Zhu, W.; Xu, Y.; Wang, N.; Han, B.; Song, H.; Qiao, J. Bioinformatic Analysis Identifies Potential Key Genes in the Pathogenesis of Turner Syndrome. Front Endocrinol. 2020, 11, 104.
- Urbach, A.; Benvenisty, N. Studying early lethality of 45,XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS ONE 2009, 4, e4175.
- Qi, X.; Wang, Q.; Yu, M.; Kong, Y.; Shi, F.; Wang, S. Bioinformatic analysis identifies the immunological profile of turner syndrome with different X chromosome origins. Front Endocrinol. 2023, 14, 1024244.
- Viuff, M.H.; Berglund, A.; Juul, S.; Andersen, N.H.; Stochholm, K.; Gravholt, C.H. Sex Hormone Replacement Therapy in Turner Syndrome: Impact on Morbidity and Mortality. J. Clin. Endocrinol. Metab. 2020, 105, 468–478.
- Gravholt, C.H.; Juul, S.; Naeraa, R.W.; Hansen, J. Morbidity in Turner syndrome. J. Clin. Epidemiol. 1998, 51, 147–158.
- Schoemaker, M.J.; Swerdlow, A.J.; Higgins, C.D.; Wright, A.F.; Jacobs, P.A.; United Kingdom Clinical Cytogenetics, G. Mortality in women with turner syndrome in Great Britain: A national cohort study. J. Clin. Endocrinol. Metab. 2008, 93, 4735–4742.
- Tuke, M.A.; Ruth, K.S.; Wood, A.R.; Beaumont, R.N.; Tyrrell, J.; Jones, S.E.; Yaghootkar, H.; Turner, C.L.S.; Donohoe, M.E.; Brooke, A.M.; et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet. Med. 2019, 21, 877–886.
- Cameron-Pimblett, A.; La Rosa, C.; King, T.F.J.; Davies, M.C.; Conway, G.S. The Turner syndrome life course project: Karyotype-phenotype analyses across the lifespan. Clin. Endocrinol. 2017, 87, 532–538.
- Bernard, V.; Donadille, B.; Zenaty, D.; Courtillot, C.; Salenave, S.; Brac de la Perriere, A.; Albarel, F.; Fevre, A.; Kerlan, V.; Brue, T.; et al. Spontaneous fertility and pregnancy outcomes amongst 480 women with Turner syndrome. Hum. Reprod. 2016, 31, 782–788.
- Denes, A.M.; Landin-Wilhelmsen, K.; Wettergren, Y.; Bryman, I.; Hanson, C. The proportion of diploid 46,XX cells increases with time in women with Turner syndrome–A 10-year follow-up study. Genet. Test Mol. Biomark. 2015, 19, 82–87.
- El-Mansoury, M.; Barrenas, M.L.; Bryman, I.; Hanson, C.; Larsson, C.; Wilhelmsen, L.; Landin-Wilhelmsen, K. Chromosomal mosaicism mitigates stigmata and cardiovascular risk factors in Turner syndrome. Clin. Endocrinol. 2007, 66, 744–751.
- Bryman, I.; Sylven, L.; Berntorp, K.; Innala, E.; Bergstrom, I.; Hanson, C.; Oxholm, M.; Landin-Wilhelmsen, K. Pregnancy rate and outcome in Swedish women with Turner syndrome. Fertil. Steril. 2011, 95, 2507–2510.
- Sybert, V.P. Phenotypic effects of mosaicism for a 47,XXX cell line in Turner syndrome. J. Med. Genet. 2002, 39, 217–220.
- Snyder, E.A.; San Roman, A.K.; Pina-Aguilar, R.E.; Steeves, M.A.; McNamara, E.A.; Souter, I.; Hayes, F.J.; Levitsky, L.L.; Lin, A.E. Genetic counseling for women with 45,X/46,XX mosaicism: Towards more personalized management. Eur. J. Med. Genet. 2021, 64, 104140.
- Luo, Y.; Zhu, D.; Du, R.; Gong, Y.; Xie, C.; Xu, X.; Fan, Y.; Yu, B.; Sun, X.; Chen, Y. Uniparental disomy of the entire X chromosome in Turner syndrome patient-specific induced pluripotent stem cells. Cell Discov. 2015, 1, 15022.
- Hovatta, O. Pregnancies in women with Turner’s syndrome. Ann. Med. 1999, 31, 106–110.
- Birkebaek, N.H.; Cruger, D.; Hansen, J.; Nielsen, J.; Bruun-Petersen, G. Fertility and pregnancy outcome in Danish women with Turner syndrome. Clin. Genet. 2002, 61, 35–39.
- Hadnott, T.N.; Gould, H.N.; Gharib, A.M.; Bondy, C.A. Outcomes of spontaneous and assisted pregnancies in Turner syndrome: The U.S. National Institutes of Health experience. Fertil. Steril. 2011, 95, 2251–2256.
- Lippe, B. Turner syndrome. Endocrinol. Metab. Clin. N. Am. 1991, 20, 121–152.
- Cleemann, L.; Holm, K.; Fallentin, E.; Skouby, S.O.; Smedegaard, H.; Moller, N.; Borch-Christensen, H.; Jeppesen, E.M.; Wieslander, S.B.; Andersson, A.M.; et al. Uterus and ovaries in girls and young women with Turner syndrome evaluated by ultrasound and magnetic resonance imaging. Clin. Endocrinol. 2011, 74, 756–761.
- Weiss, L. Additional evidence of gradual loss of germ cells in the pathogenesis of streak ovaries in Turner’s syndrome. J. Med. Genet. 1971, 8, 540–544.
- Prakash, S.K.; Crenshaw, M.L.; Backeljauw, P.F.; Silberbach, M.; Scurlock, C.; Culin, D.D.; Ranallo, K.C.; Lin, A.E. 45,X mosaicism in a population-based biobank: Implications for Turner syndrome. Genet. Med. 2019, 21, 1882–1883.
- Negreiros, L.P.; Bolina, E.R.; Guimaraes, M.M. Pubertal development profile in patients with Turner syndrome. J. Pediatr. Endocrinol. Metab. 2014, 27, 845–849.
- Viuff, M.; Gravholt, C.H. Turner Syndrome and Fertility. Ann. Endocrinol. 2022, 83, 244–249.
- Reynaud, K.; Cortvrindt, R.; Verlinde, F.; De Schepper, J.; Bourgain, C.; Smitz, J. Number of ovarian follicles in human fetuses with the 45,X karyotype. Fertil. Steril. 2004, 81, 1112–1119.
- Hovatta, O. Ovarian function and in vitro fertilization (IVF) in Turner syndrome. Pediatr. Endocrinol. Rev. 2012, 9, 713–717.
- Bakalov, V.K.; Shawker, T.; Ceniceros, I.; Bondy, C.A. Uterine development in Turner syndrome. J. Pediatr. 2007, 151, 528–531.
- Viuff, M.H.; Just, J.; Brun, S.; Dam, T.V.; Hansen, M.; Melgaard, L.; Hougaard, D.M.; Lappe, M.; Gravholt, C.H. Women with Turner Syndrome Are Both Estrogen and Androgen Deficient: The Impact of Hormone Replacement Therapy. J. Clin. Endocrinol. Metab. 2022, 107, 1983–1993.
- Hagen, C.P.; Main, K.M.; Kjaergaard, S.; Juul, A. FSH, LH, inhibin B and estradiol levels in Turner syndrome depend on age and karyotype: Longitudinal study of 70 Turner girls with or without spontaneous puberty. Hum. Reprod. 2010, 25, 3134–3141.
- Ljubicic, M.L.; Busch, A.S.; Upners, E.N.; Fischer, M.B.; Petersen, J.H.; Raket, L.L.; Frederiksen, H.; Johannsen, T.H.; Juul, A.; Hagen, C.P. A Biphasic Pattern of Reproductive Hormones in Healthy Female Infants: The COPENHAGEN Minipuberty Study. J. Clin. Endocrinol. Metab. 2022, 107, 2598–2605.
- Paterson, W.F.; Hollman, A.S.; Donaldson, M.D. Poor uterine development in Turner syndrome with oral oestrogen therapy. Clin. Endocrinol. 2002, 56, 359–365.
- Practice Committee of American Society for Reproductive Medicine. Increased maternal cardiovascular mortality associated with pregnancy in women with Turner syndrome. Fertil. Steril. 2012, 97, 282–284.
- Cauldwell, M.; Steer, P.J.; Adamson, D.; Alexander, C.; Allen, L.; Bhagra, C.; Bolger, A.; Bonner, S.; Calanchini, M.; Carroll, A.; et al. Pregnancies in women with Turner syndrome: A retrospective multicentre UK study. BJOG 2022, 129, 796–803.
- Silberbach, M.; Roos-Hesselink, J.W.; Andersen, N.H.; Braverman, A.C.; Brown, N.; Collins, R.T.; De Backer, J.; Eagle, K.A.; Hiratzka, L.F.; Johnson, W.H., Jr.; et al. Cardiovascular Health in Turner Syndrome: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000048.
- Miyabara, S.; Nakayama, M.; Suzumori, K.; Yonemitsu, N.; Sugihara, H. Developmental analysis of cardiovascular system of 45,X fetuses with cystic hygroma. Am. J. Med. Genet. 1997, 68, 135–141.
- Angelini, P. Coronary artery anomalies: An entity in search of an identity. Circulation 2007, 115, 1296–1305.
- Koenraadt, W.M.C.; Siebelink, H.J.; Bartelings, M.M.; Schalij, M.J.; van der Vlugt, M.J.; van den Bosch, A.E.; Budde, R.P.J.; Roos-Hesselink, J.W.; Duijnhouwer, A.L.; van den Hoven, A.T.; et al. Coronary anatomy in Turner syndrome versus patients with isolated bicuspid aortic valves. Heart 2019, 105, 701–707.
- Viuff, M.H.; Trolle, C.; Wen, J.; Jensen, J.M.; Norgaard, B.L.; Gutmark, E.J.; Gutmark-Little, I.; Mortensen, K.H.; Gravholt, C.H.; Andersen, N.H. Coronary artery anomalies in Turner Syndrome. J. Cardiovasc. Comput. Tomogr. 2016, 10, 480–484.
- Zakaria, D.; Tang, X.; Bhakta, R.; ElHassan, N.O.; Prodhan, P. Chromosomal Abnormalities Affect the Surgical Outcome in Infants with Hypoplastic Left Heart Syndrome: A Large Cohort Analysis. Pediatr. Cardiol. 2018, 39, 11–18.
- Phillips, H.M.; Mahendran, P.; Singh, E.; Anderson, R.H.; Chaudhry, B.; Henderson, D.J. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc. Res. 2013, 99, 452–460.
- Van Nisselrooij, A.E.L.; Lugthart, M.A.; Clur, S.A.; Linskens, I.H.; Pajkrt, E.; Rammeloo, L.A.; Rozendaal, L.; Blom, N.A.; van Lith, J.M.M.; Knegt, A.C.; et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet. Med. 2020, 22, 1206–1214.
- Sawada, H.; Rateri, D.L.; Moorleghen, J.J.; Majesky, M.W.; Daugherty, A. Smooth Muscle Cells Derived from Second Heart Field and Cardiac Neural Crest Reside in Spatially Distinct Domains in the Media of the Ascending Aorta-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1722–1726.
- Mortensen, K.H.; Hjerrild, B.E.; Andersen, N.H.; Sorensen, K.E.; Horlyck, A.; Pedersen, E.M.; Lundorf, E.; Christiansen, J.S.; Gravholt, C.H. Abnormalities of the major intrathoracic arteries in Turner syndrome as revealed by magnetic resonance imaging. Cardiol. Young 2010, 20, 191–200.
- Kim, H.K.; Gottliebson, W.; Hor, K.; Backeljauw, P.; Gutmark-Little, I.; Salisbury, S.R.; Racadio, J.M.; Helton-Skally, K.; Fleck, R. Cardiovascular anomalies in Turner syndrome: Spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. AJR Am. J. Roentgenol. 2011, 196, 454–460.
- Ghazi Sherbaf, F.; Mohajer, B.; Ashraf-Ganjouei, A.; Mojtahed Zadeh, M.; Javinani, A.; Sanjari Moghaddam, H.; Shirin Shandiz, M.; Aarabi, M.H. Serum Insulin-Like Growth Factor-1 in Parkinson’s Disease; Study of Cerebrospinal Fluid Biomarkers and White Matter Microstructure. Front. Endocrinol. 2018, 9, 608.
- Kruger, T.; Forkavets, O.; Veseli, K.; Lausberg, H.; Vohringer, L.; Schneider, W.; Bamberg, F.; Schlensak, C. Ascending aortic elongation and the risk of dissection. Eur. J. Cardiothorac. Surg. 2016, 50, 241–247.
- Patel, A.; Costello, J.M.; Backer, C.L.; Pasquali, S.K.; Hill, K.D.; Wallace, A.S.; Jacobs, J.P.; Jacobs, M.L. Prevalence of Noncardiac and Genetic Abnormalities in Neonates Undergoing Cardiac Operations: Analysis of The Society of Thoracic Surgeons Congenital Heart Surgery Database. Ann. Thorac. Surg. 2016, 102, 1607–1614.
- Mortensen, K.H.; Hjerrild, B.E.; Stochholm, K.; Andersen, N.H.; Sorensen, K.E.; Lundorf, E.; Horlyck, A.; Pedersen, E.M.; Christiansen, J.S.; Gravholt, C.H. Dilation of the ascending aorta in Turner syndrome–a prospective cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2011, 13, 24.
- Ostberg, J.E.; Donald, A.E.; Halcox, J.P.; Storry, C.; McCarthy, C.; Conway, G.S. Vasculopathy in Turner syndrome: Arterial dilatation and intimal thickening without endothelial dysfunction. J. Clin. Endocrinol. Metab. 2005, 90, 5161–5166.
- Dulac, Y.; Pienkowski, C.; Abadir, S.; Tauber, M.; Acar, P. Cardiovascular abnormalities in Turner’s syndrome: What prevention? Arch. Cardiovasc. Dis. 2008, 101, 485–490.
- Roulot, D.; Degott, C.; Chazouilleres, O.; Oberti, F.; Cales, P.; Carbonell, N.; Benferhat, S.; Bresson-Hadni, S.; Valla, D. Vascular involvement of the liver in Turner’s syndrome. Hepatology 2004, 39, 239–247.
- Corbitt, H.; Morris, S.A.; Gravholt, C.H.; Mortensen, K.H.; Tippner-Hedges, R.; Silberbach, M.; Maslen, C.L.; Gen, T.A.C.R.I. TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genet. 2018, 14, e1007692.
- Rabkin, S.W. Differential expression of MMP-2, MMP-9 and TIMP proteins in thoracic aortic aneurysm–comparison with and without bicuspid aortic valve: A meta-analysis. Vasa 2014, 43, 433–442.
- Trolle, C.; Nielsen, M.M.; Skakkebaek, A.; Lamy, P.; Vang, S.; Hedegaard, J.; Nordentoft, I.; Orntoft, T.F.; Pedersen, J.S.; Gravholt, C.H. Widespread DNA hypomethylation and differential gene expression in Turner syndrome. Sci. Rep. 2016, 6, 34220.
- Albareda, M.M.; Gallego, A.; Enriquez, J.; Rodriguez, J.L.; Webb, S.M. Biochemical liver abnormalities in Turner’s syndrome. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1037–1039.
- Floreani, A.; Molaro, M.; Baragiotta, A.; Naccarato, R. Chronic cholestasis associated with Turner’s syndrome. Digestion 1999, 60, 587–589.
- Salerno, M.; Di Maio, S.; Gasparini, N.; Rizzo, M.; Ferri, P.; Vajro, P. Liver abnormalities in Turner syndrome. Eur. J. Pediatr. 1999, 158, 618–623.
- Gardner, L.I. Letter: Intrahepatic bile stasis in 45,X Turner’s syndrome. N. Engl. J. Med. 1974, 290, 406.
- Krivosheev, A.B. Development of liver cirrhosis in a female patient with Shereshevskii-Turner syndrome. Klin. Med. 1990, 68, 95–96.
- Garavelli, L.; Donadio, A.; Banchini, G.; Fornaciari, G.; Plancher, A.C.; Franchi, F.; Gardini, G. Liver abnormalities and portal hypertension in Ullrich-Turner syndrome. Am. J. Med. Genet. 1998, 80, 180–182.
- Singh, I.; Noel, G.; Barker, J.M.; Chatfield, K.C.; Furniss, A.; Khanna, A.D.; Nokoff, N.J.; Patel, S.; Pyle, L.; Nahata, L.; et al. Hepatic abnormalities in youth with Turner syndrome. Liver Int. 2022, 42, 2237–2246.
- Machlab, S.; Miquel, M.; Volta, T.; Escoda, M.R.; Vergara, M. Turner syndrome as a cause of liver cirrhosis. Gastroenterol. Hepatol. 2018, 41, 308–309.
- El-Mansoury, M.; Berntorp, K.; Bryman, I.; Hanson, C.; Innala, E.; Karlsson, A.; Landin-Wilhelmsen, K. Elevated liver enzymes in Turner syndrome during a 5-year follow-up study. Clin. Endocrinol. 2008, 68, 485–490.
- Larizza, D.; Locatelli, M.; Vitali, L.; Vigano, C.; Calcaterra, V.; Tinelli, C.; Sommaruga, M.G.; Bozzini, A.; Campani, R.; Severi, F. Serum liver enzymes in Turner syndrome. Eur. J. Pediatr. 2000, 159, 143–148.
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Engl. J. Med. 2004, 351, 1227–1238.
- Izumita, Y.; Nishigaki, S.; Satoh, M.; Takubo, N.; Numakura, C.; Takahashi, I.; Soneda, S.; Abe, Y.; Kamasaki, H.; Ohtsu, Y.; et al. Retrospective study of the renal function using estimated glomerular filtration rate and congenital anomalies of the kidney-urinary tract in pediatric Turner syndrome. Congenit. Anom. 2020, 60, 175–179.
- Gravholt, C.H.; Andersen, N.H.; Conway, G.S.; Dekkers, O.M.; Geffner, M.E.; Klein, K.O.; Lin, A.E.; Mauras, N.; Quigley, C.A.; Rubin, K.; et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur. J. Endocrinol. 2017, 177, G1–G70.
- Ogawa, T.; Takizawa, F.; Mukoyama, Y.; Ogawa, A.; Ito, J. Renal morphology and function from childhood to adulthood in Turner syndrome. Clin. Exp. Nephrol. 2021, 25, 633–640.
- Egli, F.; Stalder, G. Malformations of kidney and urinary tract in common chromosomal aberrations. I. Clinical studies. Humangenetik 1973, 18, 1–15.
- Gregoir, W. Conservative Surgery in Horseshoe-Kidney. Urol. Int. 1963, 16, 129–138.
- Pritti, K.; Mishra, V.; Patel, H. A Rare Case of Mosaic Ring Turner Syndrome with Horseshoe Kidney. J. Hum. Reprod. Sci. 2022, 15, 318–320.
- Bilge, I.; Kayserili, H.; Emre, S.; Nayir, A.; Sirin, A.; Tukel, T.; Bas, F.; Kilic, G.; Basaran, S.; Gunoz, H.; et al. Frequency of renal malformations in Turner syndrome: Analysis of 82 Turkish children. Pediatr. Nephrol. 2000, 14, 1111–1114.
- Horita, S.; Simsek, E.; Simsek, T.; Yildirim, N.; Ishiura, H.; Nakamura, M.; Satoh, N.; Suzuki, A.; Tsukada, H.; Mizuno, T.; et al. SLC4A4 compound heterozygous mutations in exon-intron boundary regions presenting with severe proximal renal tubular acidosis and extrarenal symptoms coexisting with Turner’s syndrome: A case report. BMC Med. Genet. 2018, 19, 103.
- Cintron, D.; Rodriguez-Gutierrez, R.; Serrano, V.; Latortue-Albino, P.; Erwin, P.J.; Murad, M.H. Effect of estrogen replacement therapy on bone and cardiovascular outcomes in women with turner syndrome: A systematic review and meta-analysis. Endocrine 2017, 55, 366–375.
- Gravholt, C.H. Aspects of the treatment of Turner syndrome. Expert Opin. Pharmacother. 2001, 2, 1633–1647.
- Gussinye, M.; Terrades, P.; Yeste, D.; Vicens-Calvet, E.; Carrascosa, A. Low areal bone mineral density values in adolescents and young adult turner syndrome patients increase after long-term transdermal estradiol therapy. Horm. Res. 2000, 54, 131–135.
- Elsheikh, M.; Conway, G.S.; Wass, J.A. Medical problems in adult women with Turner’s syndrome. Ann. Med. 1999, 31, 99–105.
- Saenger, P.; Wikland, K.A.; Conway, G.S.; Davenport, M.; Gravholt, C.H.; Hintz, R.; Hovatta, O.; Hultcrantz, M.; Landin-Wilhelmsen, K.; Lin, A.; et al. Recommendations for the diagnosis and management of Turner syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 3061–3069.
- Landin-Wilhelmsen, K.; Bryman, I.; Windh, M.; Wilhelmsen, L. Osteoporosis and fractures in Turner syndrome-importance of growth promoting and oestrogen therapy. Clin. Endocrinol. 1999, 51, 497–502.
- Even, L.; Bronstein, V.; Hochberg, Z. Bone maturation in girls with Turner’s syndrome. Eur. J. Endocrinol. 1998, 138, 59–62.
- Holroyd, C.R.; Davies, J.H.; Taylor, P.; Jameson, K.; Rivett, C.; Cooper, C.; Dennison, E.M. Reduced cortical bone density with normal trabecular bone density in girls with Turner syndrome. Osteoporos Int. 2010, 21, 2093–2099.
- Wasserman, H.; Backeljauw, P.F.; Khoury, J.C.; Kalkwarf, H.J.; Gordon, C.M. Bone fragility in Turner syndrome: Fracture prevalence and risk factors determined by a national patient survey. Clin. Endocrinol. 2018, 89, 46–55.
- Bakalov, V.K.; Chen, M.L.; Baron, J.; Hanton, L.B.; Reynolds, J.C.; Stratakis, C.A.; Axelrod, L.E.; Bondy, C.A. Bone mineral density and fractures in Turner syndrome. Am. J. Med. 2003, 115, 259–264.
- Soucek, O.; Lebl, J.; Snajderova, M.; Kolouskova, S.; Rocek, M.; Hlavka, Z.; Cinek, O.; Rittweger, J.; Sumnik, Z. Bone geometry and volumetric bone mineral density in girls with Turner syndrome of different pubertal stages. Clin. Endocrinol. 2011, 74, 445–452.
- Bakalov, V.K.; Axelrod, L.; Baron, J.; Hanton, L.; Nelson, L.M.; Reynolds, J.C.; Hill, S.; Troendle, J.; Bondy, C.A. Selective reduction in cortical bone mineral density in turner syndrome independent of ovarian hormone deficiency. J. Clin. Endocrinol. Metab. 2003, 88, 5717–5722.
- Reiss, A.L.; Mazzocco, M.M.; Greenlaw, R.; Freund, L.S.; Ross, J.L. Neurodevelopmental effects of X monosomy: A volumetric imaging study. Ann. Neurol. 1995, 38, 731–738.
- Murphy, D.G.; DeCarli, C.; Daly, E.; Haxby, J.V.; Allen, G.; White, B.J.; McIntosh, A.R.; Powell, C.M.; Horwitz, B.; Rapoport, S.I.; et al. X-chromosome effects on female brain: A magnetic resonance imaging study of Turner’s syndrome. Lancet 1993, 342, 1197–1200.
- Reiss, A.L.; Freund, L.; Plotnick, L.; Baumgardner, T.; Green, K.; Sozer, A.C.; Reader, M.; Boehm, C.; Denckla, M.B. The effects of X monosomy on brain development: Monozygotic twins discordant for Turner’s syndrome. Ann. Neurol. 1993, 34, 95–107.
- Kesler, S.R.; Blasey, C.M.; Brown, W.E.; Yankowitz, J.; Zeng, S.M.; Bender, B.G.; Reiss, A.L. Effects of X-monosomy and X-linked imprinting on superior temporal gyrus morphology in Turner syndrome. Biol. Psychiatry 2003, 54, 636–646.
- Lepage, J.F.; Mazaika, P.K.; Hong, D.S.; Raman, M.; Reiss, A.L. Cortical brain morphology in young, estrogen-naive, and adolescent, estrogen-treated girls with Turner syndrome. Cereb. Cortex 2013, 23, 2159–2168.
- Marzelli, M.J.; Hoeft, F.; Hong, D.S.; Reiss, A.L. Neuroanatomical spatial patterns in Turner syndrome. Neuroimage 2011, 55, 439–447.
- Knickmeyer, R.C. Turner syndrome: Advances in understanding altered cognition, brain structure and function. Curr. Opin. Neurol. 2012, 25, 144–149.
- Heard, E.; Chaumeil, J.; Masui, O.; Okamoto, I. Mammalian X-chromosome inactivation: An epigenetics paradigm. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 89–102.
- Sahakyan, A.; Kim, R.; Chronis, C.; Sabri, S.; Bonora, G.; Theunissen, T.W.; Kuoy, E.; Langerman, J.; Clark, A.T.; Jaenisch, R.; et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem. Cell 2017, 20, 87–101.
- Berletch, J.B.; Yang, F.; Disteche, C.M. Escape from X inactivation in mice and humans. Genome. Biol. 2010, 11, 213.
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes that escape from X inactivation. Hum. Genet. 2011, 130, 237–245.
- Backeljauw, P.; Chernausek, S.D.; Gravholt, C.H.; Kruszka, P. Turner syndrome. In Sperling Pediatric Endocrinology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 627–660.
- Jones, M.H.; Furlong, R.A.; Burkin, H.; Chalmers, I.J.; Brown, G.M.; Khwaja, O.; Affara, N.A. The Drosophila developmental gene fat facets has a human homologue in Xp11.4 which escapes X-inactivation and has related sequences on Yq11.2. Hum. Mol. Genet. 1996, 5, 1695–1701.
- Quilter, C.R.; Karcanias, A.C.; Bagga, M.R.; Duncan, S.; Murray, A.; Conway, G.S.; Sargent, C.A.; Affara, N.A. Analysis of X chromosome genomic DNA sequence copy number variation associated with premature ovarian failure (POF). Hum. Reprod. 2010, 25, 2139–2150.
- Laupacis, A.; Bourne, R.; Rorabeck, C.; Feeny, D.; Tugwell, P.; Wong, C. Comparison of total hip arthroplasty performed with and without cement: A randomized trial. J. Bone Joint Surg. Am. 2002, 84, 1823–1828.
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013, 34, 108–110.
- Alvarez-Nava, F.; Lanes, R. Epigenetics in Turner syndrome. Clin. Epigenetics 2018, 10, 45.