 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mathilde Vermeer | -- | 2947 | 2022-10-19 16:14:44 | | | |
| 2 | Amina Yu | + 5022 word(s) | 7969 | 2022-10-20 03:49:33 | | |
Video Upload Options
Desmosomes are mirroring, transmembrane protein chains that connect the intermediate filament networks of neighbouring cells. Each chain continuously (dis)assembles due to the turnover of five desmosomal protein types: desmoplakin, plakoglobin, plakophilins, desmocollins and desmogleins. The expression of two genes is critical to the formation of all desmosomes: namely DSP, encoding two differently spliced desmoplakin proteins (DPI and DPII) and JUP, encoding plakoglobin (PG). Meanwhile, plakophilins, desmocollins and desmogleins are expressed in a tissue-specific manner and are therefore encoded by multiple genes.
1. Introduction
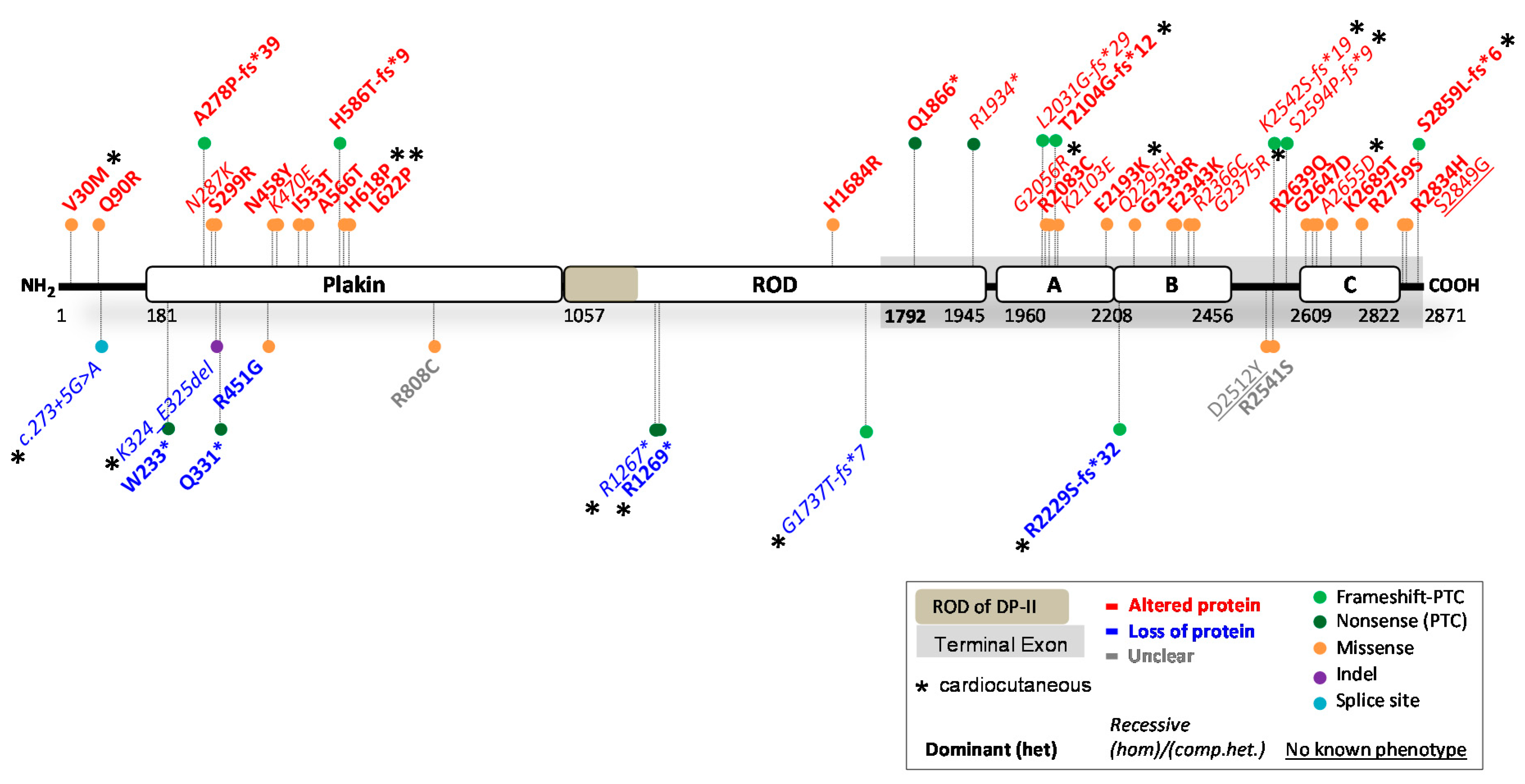
2. Reported DSP Variants
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) | Protein Domain |
ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect | Prediction: Functional | Skin | Heart |
|---|---|---|---|---|---|---|---|---|---|
| c.88G > A | p.(Val30Met) | N-terminus | B | >Protein expressed -PolyPhen-2> Benign (0.000) -SIFT> NOT tolerated -MutPred2> Benign (0.092) |
Altered DP function; Mutant DP protein expressed, normal size and amount (WB) [9][10][11]. |
Binding to PG abolished (Co-IP); DP localization in cytoplasm (transfection) [9]; DP normal in myocardial and epidermal tissue. Exhibit weaker binding to iASPP (transfection) [11]. Mouse DSPWT/88G > A [9]. |
Match | (het) PPK; (het) WH2 | (het) ACM |
| c.269A > G | p.(Gln90Arg) | N-terminus | B | >Protein expressed -PolyPhen-2> Probably damaging (0.967) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.757) |
Altered DP function; Mutant DP protein expressed, normal size and amount (WB) [9][11]. |
Binding to PG abolished (Co-IP); DP localization to cytoplasm (transfection) [9]. Mouse DSPWT/269A > G [9] |
Match | n.s. | (het) ACM |
| c.273 + 5G > A | Multiple splice products |
Intron splice site (N-terminus) |
US | -Human Splicing Finder> Broken WT donor site -MaxEntScan> Alteration of WT donor site, probably affecting splicing > Altered splicing, out of frame> PTC > NMD |
Partial loss of DP: 20% less DP product on WB. No alternatively spliced transcripts discovered in patient-derived cells ([12][13], but did so in in vitro splicing assay (transfection). However, not functional [13]. |
-In combination with c.6687del> Reduced DP protein on blot and staining in explanted heart and hiPSC-CMs and primary KCs [13][14]. -Dislocation of DP after 2D mechanical stretch; resulted in reduced count and density of desmosomes (EM) in dynamic EHTs leading to lower force and stress [14]. |
Partial match, normal splicing left. |
(comp.het) PPK; WH with DSP: c.6687delA |
(comp.het) ACM/ NCCM :with DSP: c.6687delA |
| c.699G > A | p.(Trp233*) | Plakin-domain | P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.55385) |
Partial loss of DP; Mutant RNA not detected in patient cells. Mutant DP is unstable (transfection-WB) [9]. |
Perinuclear aggregates of DP (transfection IF) [9]. | Partial match, but not with transfection IF/WB | n.s. | (het) ACM |
| c.832del | p.(Ala278Pro fs*39) |
Plakin-domain | P | >PTC > NMD, no protein | Altered DP function; Truncated DP normally expressed and protein runs at 60 kDa (315 aa) (transfection-WB) -Leads to truncated DSP mRNA, also indicating that mRNA translation following the truncation was completely impaired. |
c.832del overexpression led to upregulation of PG and downregulation of β-catenin in the nuclei, without affecting their expression in the cytoplasm (transfection) [15]. | Mismatch | n.s. | (het) ACM |
| c.861T > G | p.(Asn287Lys) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.997) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.699) |
Altered DP function; Mutant DP expressed [16]. |
Aberrant DP and Cx43 localization (transfection-IF) [16]. | Match | (hom)PPK; (hom)WH; (hom)EBS | n.o. |
| c.897C > G | p.(Ser299Arg) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.672) |
Altered DP function; Mutant DP expressed [11]. |
Exhibit weaker binding to iASPP = desmosome regulator (transfection) [11]. | Match | n.s. | (het) ACM |
| c.939 + 1G > A | p.(Gln331*) | Donor site intron 7 (Plakin-domain) |
P | >PTC > NMD, no protein | Partial loss of DP; Absence of detection of mRNA in multiple patient KCs, reported by two studies, suggests efficient NMD [17][18]. Only 20% DPI and 50% DPII is left on WB [19]. |
-Major abnormalities in the spinous layer of the epidermis. The intercellular space is widened and KCs contain abnormal cytoplasmic densities [18]. -Small desmosomes and fewer in number; perinuclear keratin distribution 7. - DC3 seems reduced on WB; volume densities of desmosomal proteins seem different from control [19]. |
Match | (het) PPK | n.r. |
| c.969_974del | p.(Lys324_Glu325del) | Plakin-domain | LP | >Protein expressed -MutPred-Indel> Benign (0.19566) |
Partial loss of DP; Reduced expression of both native DP isoforms in cytoskeletal and cytoplasmic protein fractions (WB) [10], suggesting instable protein> incomplete degradation. | DP expression was significantly reduced in myocardial tissue and epidermal biopsies (IF) [10]. | Mismatch | (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular |
| c.1348C > G | p.(Arg451Gly) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.756) |
Partial loss of DP; 50% reduced DPI&II protein in EHTs (WB) [20]. -mRNA levels of DSP not reduced compared to WT [20]. > instable protein degradation. |
50% reduced DP signal and 70% reduced Cx43 in myocardial tissues (IF); Proteolytic degradation by calpain, leading to DP insufficiency [20]. | Mismatch | n.r. | (het) ACM, bi-ventricular |
| c.1372A > T | p.(Asn458Tyr) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Possibly damaging (0.939) -SIFT> Tolerated -MutPred2> Benign (0.323) |
Altered DP function; Mutant DP expressed [16]. |
Altered EB1 binding and Cx43 localization (transfection IF) [16]. | Match | n.o. | (het) ACM |
| c.1408A > G | p.(Lys470Glu) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Benign (0.082) -SIFT> Tolerated -MutPred2> Benign (0.408) |
Altered DP function; Conformational alternation, but overall folded structure of DP is remained [21]. Mutant DP expressed (WB) [16]. |
Mutant is incorporated into the desmosome [10]. | Match | n.s./n.o. | (hom) ACM |
| c.1598T > C | p.(Ile533Thr) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Probably damaging (0.998) -SIFT> NOT tolerated -MutPred2> Benign (0.442) |
Altered DP function; Mutant DP expressed (WB) [16]. |
Altered EB1 binding and Cx43 localization (transfection IF) [16]. | Match | n.o. | (het) ACM |
| c.1696G > A | p.(Ala566Thr) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Benign (0.007) -SIFT> Tolerated -MutPred2> Benign (0.153) |
Altered DP function; Mutant DP expressed (WB) [16]. |
Mutant is incorporated into the desmosome [10]. | Match | n.o. | (het) ACM |
| c.1853A > C | p.(His618Pro) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Possibly damaging (0.602) -SIFT> NOT tolerated -MutPred2> Benign (0.540) |
Altered DP function; Mutant DP expressed (WB) [22]. |
Mutant localizes to membrane, affected Cx43 localization (transfection studies/skin biopsies). Desmosome aggregation [22]. | Match | (het) PPK; (het) WH; (het) EBS | (het) CM |
| c.1865T > C | p.(Leu622Pro) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.998) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.828) |
Altered DP function; Mutant DP expressed (WB) [22]. |
Mutant localizes to membrane, affected Cx43 localization (transfection studies/skin biopsies). Desmosome aggregation [22]. | Match | (het) PPK; (het) WH; (het) EBS | (het) CM |
| c.1755dup | p.(His586Thr fs*9) |
Plakin domain |
P | >PTC > NMD, no protein | Altered DP function; Truncated DP protein, (65 kDa) (WB), truncation of ROD and C-terminus [23]. |
n.r. | Mismatch | n.s. | (het) ACM, LV mostly |
| c.2422C > T | p.(Arg808Cys) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Benign (0.047) -SIFT> NOT tolerated -MutPred2> Benign (0.409) |
Unclear; Conformational alteration (transfection), but overall folded structure of DP is remained [21]. Needs further confirmation in patient cells, whether expressed or not. |
n.r. | Unclear | n.s. | (het) ACM |
| c.3799C > T | p.(Arg1267*) | ROD domain (DPI) |
P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.5552) |
Partial loss of DPI; Instable mutant DPI protein (NMD?) [24]. -Highly reduced DSP mRNA expression (NMD?) [24]. -Complete loss of DPI in patient skin, DPII has normal expression as expected (WB) [24]. |
n.r. | Match | (hom)PPK epidermolytic; (hom)WH |
(hom) ACM/ DCM |
| c.3805C > T | p.(Arg1269*) | ROD domain (DPI) |
P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.55487) |
Partial loss of DPI; Broken down by NMD. DPI/DPI-II protein ratios lower in variant carriers compared with WT individuals. -DPI/DPII expression ratio reduced by 28% in mutant cells. 15-fold lower mutant than WT [10]. |
Decreased DP expression in endomyocardial biopsies. DPI deficiency (IF) [10]. | Match | (het) PPK; (het) WH | (het) DCM, bi-ventricular |
| c.5051A > G | p.(His1684Arg) | ROD domain (DPI) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.956) -SIFT> NOT tolerated -MutPred2> Benign (0.256) |
Altered DPI function: No effect on amount or size of DPI protein on WB [25]. DPII should not be affected. |
Affects action potential and duration; multiple ion channel dysfunction in hiPSC-CMs [25]. | Match | n.r. | (het) CM, conduction disease |
| c.5208_5209del | p.(Gly1737Thrfs*7) | ROD domain (DPI) |
P | >PTC > NMD, no protein | Partial loss of DPI/Unclear? Truncated DPI protein predicted to run at similar height as DPII, yet no increase in this band was observed in skin biopsies (WB) [26]. DPII should not be affected, but data are unclear. |
n.r. | Unclear | (hom) PPK acantholytic; (hom)WH; |
(hom) NCCM, bi-ventricular, severe |
| c.5596C > T | p.(Gln1866*) | ROD domain (DPI) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DPI function; Truncated DPI protein (160 kDa) observed in skin biopsies [27]. DPII should not be affected. |
n.r. | Match | n.s. | (het) ACM, LV dilation |
| c.5800C > T | p.(Arg1934*) | ROD domain (DPI) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DPI function; Truncated DPI protein (243 kDa) (WB) [28]. DPII should not be affected. -Aberrant mRNA transcripts. Not NMD. |
Stable expressed DP protein, which is recruited into desmosomes, although more punctate staining was observed (IF) [28]. | Match | (comp.het) (lethal) EBS, PPK and WH, with DSP: c.6091_6092del [28] |
n.r. |
| c.6091_6092del | p.(Leu2031Glyfs*29) | PRD (A domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Truncated DP-I protein (228 kDa) (WB) [28]. Not clear what happens with DPII. -Aberrant mRNA transcripts. Not NMD. |
Stable expressed DP protein, which is recruited in desmosomes, although more punctate staining was observed (IF) [28]. | Match | (comp.het) (lethal) EBS, PPK; and WH, with DSP: c.5800C > T [28] |
n.r. |
| c.6166G > C | p.(Gly2056Arg) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.872) |
Altered DP function; Expressed in insoluble fraction of bacterial cells (transfection WB) [29]. Low expression in HeLa cells. Likely expressed mutant. |
n.r. | Probable match | (hom) PPK | (hom) ACM, LV involvement |
| c.6247C > T | p.(Arg2083Cys) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Benign (0.443) |
Altered DP function; Expressed in soluble fraction of bacterial cells (transfection WB), thus correctly folded [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) LQTS |
| c.6307A > G | p.(Lys2103Glu) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.860) -SIFT> Tolerated -MutPred2> Benign (0.417) |
Altered DP function; Expressed in soluble fraction of bacterial cell transfection, thus correctly folded (WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) DCM |
| c.6310del | p.(Thr2104Glnfs*12) | PRD (A domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Several truncated DP proteins shown on WB, but mutant is predicted to be 238 kDa [30] |
Fibrosis and fat deposition in the heart with reduction in Cx43, disorganized IDs, but staining of DP, PG and DG2 seemed normal; severe reduction of DPI&II on IF ex vivo skin. β-catenin expression was also reduced on IF in skin [30]. | Match | (comp.het) EBS, PPK and WH: with DSP: c.7964C > A |
(comp.het) Bi-ventricular CM: with DSP: c.7964C > A |
| c.6577G > A | p.(Glu2193Lys) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.950) -SIFT> Tolerated -MutPred2> Benign (0.346) |
Altered DP function; Expressed in insoluble fraction in bacterial cells (transfection WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | (comp.het) Alopecia PPK, with DSP:c.7567delAAGA |
(comp.het) DCM, with DSP:c.7567delAAGA |
| c.6687del | p.(Arg2229Serfs*32) | PRD (B domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Partial loss of DP: NMD of product (WB, NMD inhibitor exp.), 50% reduced protein levels [13][14]. -mRNA 50% reduced [14]. |
-Reduced DP protein on blot and staining in explanted heart, hiPSC-CMs and primary KCs [13][14]. -Mislocalisation of DP after 2D mechanical stretch; in combination with c.273 + 5G > A resulted in reduced count and density of desmosomes in hiPSC-derived dynamic EHTs leading to lower force and stress [14] -Faster differentiation observed in primary KCs of patients. Mechanical stretch provoked cell-contact defects [13]. |
Mismatch NMD active in terminal exon! |
(het)PPK; (comp.het)WH: with DSP: c.273 + 5G > A |
Lethal ACM/ NCCM (comp.het) ACM/ NCCM (bi-ventricular) (het) |
| c.6885A > T | p.(Gln2295His) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.833) |
Altered DP function; Likely truncated DP protein expressed. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. | Probable match | n.r. | (hom) DCM |
| c.7012G > A | p.(Gly2338Arg) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.910) |
Altered DP function; Insoluble fraction in bacterial cell transfection (WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) CM |
| c.7027G > A | p.(Glu2343Lys) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Benign (0.077) -SIFT> Tolerated -MutPred2> Benign (0.386) |
Altered DP function; Likely truncated DP protein expressed. Soluble fraction in bacterial cell transfection, thus correctly folded (WB) [29]. |
Altered binding with vimentin and keratin8/18 (transfection IF) [31]. | Probable match | n.r. | (het) ACM ACM, with PKP2:c.1468C > T |
| c.7096C > T | p.(Arg2366Cys) | PRD (B domain) |
LP | >Protein expressed -PolyPhen-2> Probably damaging (0.980) -SIFT> NOT tolerated -MutPred2> Benign (0.622) |
Altered DP function; Likely truncated DP protein expressed [31]. Soluble fraction of bacterial cell transfection (WB) [29]. High expression in HeLa cells. Needs confirmation in patient cells. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. No binding deficiency with vimentin (IF transfection) [29]. |
Probable match | (hom)EBS; (hom)PPK; (hom)WH | n.r. |
| c.7123G > C | p.(Gly2375Arg) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.938) |
Altered DP function; Truncated DP protein expressed [32]. Insoluble fraction of bacterial cell transfection (WB) [29]. |
Co-alignment with IFs severely affected. Diffuse cytosolic distributed [32]. Targeting to IFs affected (transfection-IF) [29]. |
Match | (hom)EBS; (hom)PPK; (hom)WH | (hom)ACM |
| c.7534G > T | p.(Asp2512Tyr) | Linkers | US | >Protein expressed -PolyPhen-2> Probably damaging (0.998) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.825) |
Unclear; Likely truncated DP protein expressed. Needs further confirmation in patient cells. |
No binding deficiency with IFs (transfection-IF) [31]. | Unclear | n.o. | n.o. |
| c.7623G > T | p.(Arg2541Ser) | Linkers | US | >Protein expressed -PolyPhen-2> Benign (0.010) -SIFT> NOT tolerated -MutPred2> Benign (0.265) |
Unclear; Likely truncated DP protein expressed. Needs further confirmation in patient cells. |
No binding deficiency with IFs (transfection-IF) [31]. | Unclear | n.r. | (het) ACM |
| c.7623del | p.(Lys2542Serfs*19) | Linkers | LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Severe reduction of both DPI&II (WB), both truncated proteins detected [10]. |
Normal DP immunoreactivity in epidermal and myocardial tissue (IF)/or almost no signal depending on homozygous or heterozygous patient [10]. | Match | (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular |
| c.7780del | p.(Ser2594Profs*9) | Linkers | LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Truncated DP protein, 18 aa downstream of deletion (WB) [33]. |
Partial disruption with intermediate filament binding (IF) [33]; KCs have alteration in morphology, elasticity, adhesion capabilities and viscoelastic properties [34][35]. | Match | (hom)PPK; (hom)WH | (hom) DCM |
| c.7916G > A | p.(Arg2639Gln) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.978) -SIFT > Tolerated -MutPred2> Benign (0.484) |
Altered DP function; Likely truncated DP proteins expressed [31]. Expressed in soluble fraction in bacterial cells (transfection WB) [29]. |
Altered binding with desmin and keratin8/18 (transfection IF) [31]. No binding deficiency with vimentin (IF transfection) [29]. |
Probable match | n.r. | (het) CM; RV dysfunction |
| c.7940G > A | p.(Gly2647Asp) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.980) -SIFT> NOT tolerated -MutPred2> Benign (0.619) |
Altered DP function: Both in insoluble and soluble fraction in bacterial cells (transfection WB) [29]. Likely expressed mutant. |
n.r. | Probable match | n.r. | (het) CM |
| c.7964C > A | p.(Ala2655Asp) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.754) |
Altered DP function; Likely truncated DP proteins expressed, as full loss of protein is not expected due to recessive inheritance. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. | Probable match | (hom)EBS; (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular |
| c.8066A > C | p.(Lys2689Thr) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT > Tolerated -MutPred2> Benign (0.625) |
Altered DP function: Expressed in soluble fraction in bacterial cells (transfection WB), thus correctly folded [29]. High expression in HeLa cells. Likely expressed mutant. Needs confirmation in patient cells. |
No binding deficiency with vimentin (transfection IF) [29]. | Probable match | n.r. | (het) ACM |
| c.8275C > A | p.(Arg2759Ser) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT > Tolerated -MutPred2> Benign (0.350) |
Altered DP function: Expressed in soluble fraction in bacterial cells (transfection, thus correctly folded WB) [29]. Likely expressed mutant. Needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) ACM |
| c.8501G > A | p.(Arg2834His) | C- terminus |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.972) -SIFT> NOT tolerated -MutPred2> Benign (0.189) |
Altered DP function; C-terminally truncated DP protein (WB) [9]. |
Aberrant IF localization; DP localization at cell membrane; affects other junctional proteins [9]; Arg2834His blocked the GSK3β phosphorylation cascade and reduced DP–GSK3β interactions in KCs and in hearts of Arg2834His DP mice [2]. Mouse DSPWT/8501G > A [9][36][37] |
Match | n.s. | (het) ACM |
| Engineered variant | p.(Ser2849Gly) | C- terminus |
n.a. | >Protein expressed -PolyPhen-2> Probably damaging (0.978) -SIFT> NOT tolerated -MutPred2> Benign (0.344) |
Altered DP function; Mutant DP protein detected (WB), normal size. |
Mutant DP exhibits increased anchorage of keratin/desmin [38] filaments and fosters calcium independency [39]. | n.a. | unknown | unknown |
| c.8576_8577del | p.(Ser2859Leufs*6) | C- terminus |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Highly reduced mutant DP protein detected in insoluble fraction (WB), none in soluble fraction, but normal size (only 2859 + 6 aa, compared to wildtype 2871 aa) |
GSK3β, normally phosphorylates Ser2859Leu, translocated to the soluble fraction of patient extract where its high activity (dephosph). Ser9 was associated with the phosphorylation (Ser33/37-Thr41) and degradation of β-catenin; abolition of β-catenin phosphorylation in the non-soluble fraction was associated with its translocation into CMs nuclei [40]. | Match | (hom and het) EBS | (hom and het) ACM |
2.1. DSP Variants Causing DP Reduction
2.2. DSP Variants Causing an Altered DP Protein
2.3. Potential Therapeutic Avenues
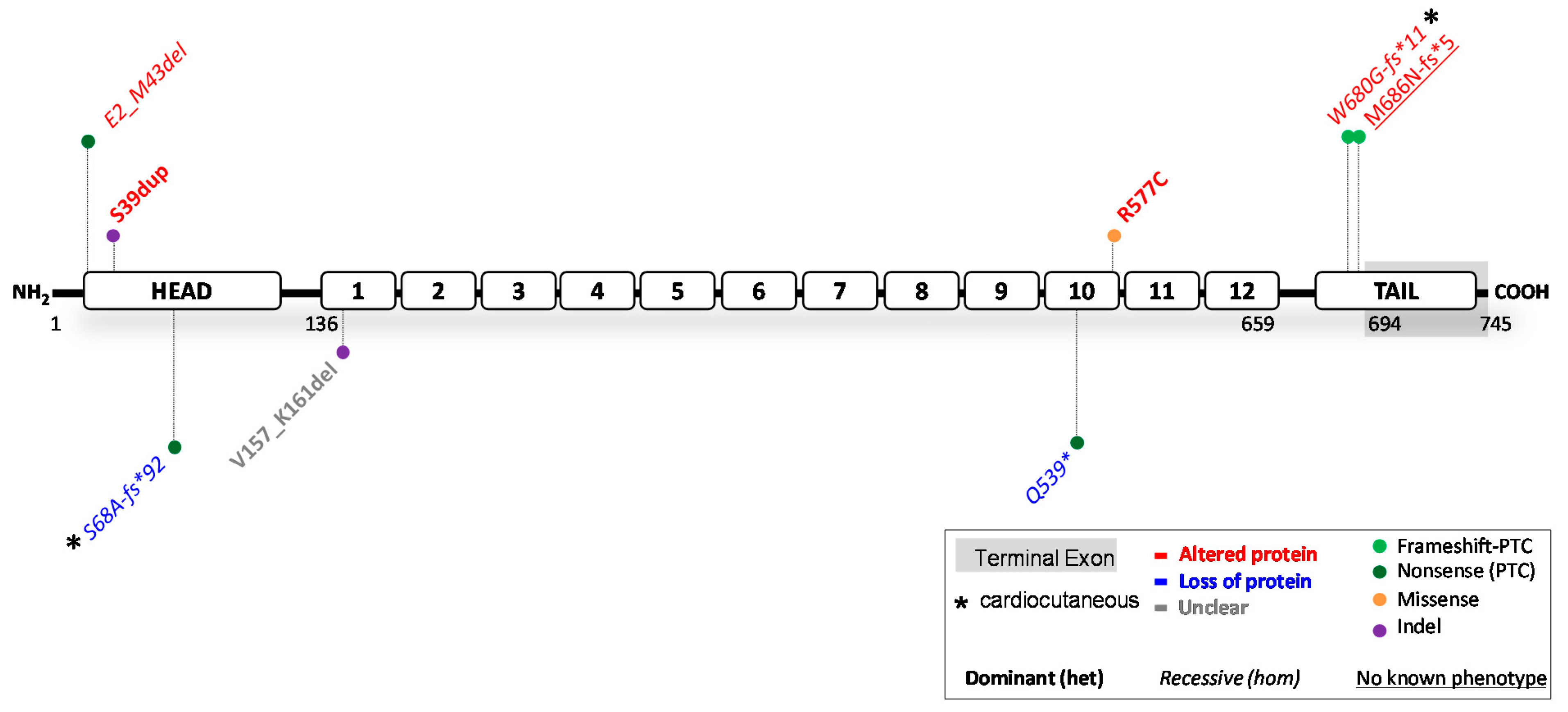
3. Reported JUP Variants
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) |
Protein Domain |
ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect | Prediction: Functional | Skin | Heart |
|---|---|---|---|---|---|---|---|---|---|
| c.71C > A | p.(Glu2_Met43del) not p.(Ser24*) as predicted |
Head domain |
LP | >PTC > NMD, no protein -MutPred-LOF> too short sequence for prediction |
Altered PG function; Truncated N-terminal protein (lacking the first 42 aa, translation re-initiation Met43) with reduced expression in the skin (WB) [49]. -Similar JUP mRNA levels as in control [49]. |
Reduced PG expression (IF; WB) and disrupted distribution of DP and DG1 (IF) [49]. | Mismatch | (hom)EBS; (hom)PPK; (hom)WH | n.o. |
| c.116_118dup | p.(Ser39dup) | Head domain |
US | >Protein expressed -MutPred-Indel> Pathogenic (0.76492) |
Altered PG function: Mutant PG protein size similar as WT (82 kDa; WB) [50][51]. See comment on biological effect [51]. |
-Patient heart displayed a decrease in signal of DP, PG and Cx43 (IF ) [50] - Transfection of HEK293 with mutant construct showed increased size of PG (90 kDa) due to ubiquitin binding (WB); cytoplasmic localization of mutant-PG (IF); higher proliferation and lower apoptosis; fewer and smaller desmosomes in mutant PG cells (EM ) [50] -Additional binding properties of mutant PG to TAIP-2 and HRC-BP (Co-IP and yeast-two-hybrid) [50] - Diminished cell stiffness, but not cell adhesion [51]. |
Match | n.o. | (het) ACM |
| c.201del | p.(Ser68Ala fs*92) |
Head domain |
P | >PTC > NMD, no protein | Loss of PG?: Highly reduced levels of JUP mRNA (normal splicing), no WB performed [52] |
-Absence of PG protein staining in skin biopsies (IF); small desmosomes and wide intercellular spaces (EM) [52]. | Match | (hom)EBS; (hom)PPK; (hom) alopecia |
(hom) ACM |
| c.469-8_469 -1del |
p.(Val157_Lys161del) | Armadillo Domain 1 |
LP | >Protein expressed -MutPred-Indel> Pathogenic (0.78939) |
Unclear; 15 nucleotides shorter cDNA fragments when compared to controls, no WB performed [53]. |
Cryptic splice acceptor site activation in exon 4 [53]. | Unclear | n.r. | (het) ACM |
| c.1615C > T | p.(Gln539*) | Armadillo Domain 10 |
P | >PTC > NMD, no protein - MutPred-LOF> Borderline pathogenic (0.62246) |
Loss of PG; No truncated, or full-length PG protein detected in patient’s skin extracts (WB) [54]. -Apart from the strong reduction of JUP (90% reduction), DSP and DSG1 mRNAs were also markedly decreased [54]. |
-Complete loss of PG protein in the patient’s skin (WB and IF, both with N-terminal and C-terminal antibody); No skin barrier formation; significant reduction of DP and DG3 in patient skin (IF). Only few, abnormal desmosomes were formed [54]. - Strong reduction PG in the myocardium [54]. |
Match | (hom) lethal EBS |
n.o. at young age |
| c.1729C > T | p.(Arg577Cys) | Armadillo Domain 10 |
LP | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> Tolerated -MutPred2> Pathogenic (0.720) |
Altered PG function: Mutant PG had similar size as WT and was not reduced on blot according to the study [55]. |
-On WB, DG2 and Cx43 protein levels were reduced in mutant expression cells, and desmosomal junctions were destabilized (transfection studies) [55]. | Match | n.r. | (het) ACM |
| c.2038_2039del | p.(Trp680Gly fs*11) |
Tail domain |
P | >PTC > NMD, no protein | Altered PG function; C-terminal truncated PG protein is abundantly expressed (56 aa missing) (WB of biopsied LV and RV of multiple patients) [51][56][57]. |
-Reduced Cx43 and PG in patient ventricles and absence of phosphorylated Cx43 (IF; WB). A decreased number of gap junctions in patient’s myocardium (EM) [57]. - Diminishes cell adhesion, but not the cell compliance [51]. -Mouse JUP knockin c.2038_2039del [58]. -The mouse data contradict the human data and suggest that mutant mRNA is broken down by NMD in mice, and not much protein is produced (WB does show truncated protein). The authors reason that although the deletion is located in exon 11, the PTC is located in the terminal exon (exon 12) > homozygous mice die at postnatal day 1, while cardiac development went normal, mice had severe skin fragility. -Fusion of the last 5 exons in mice, produced the truncated protein fully, did not cause lethality; however, mice did not develop cardiac dysfunction at 11 months of age. |
Mismatch Mismatch human: mice |
(hom)PPK; (hom)WH [59] | (hom) ACM [59] |
| c.2057_2058del | p.(Met686Asn fs*5) |
Tail domain |
n.a. | >PTC > NMD, no protein | Altered PG function; Transfected myocytes showed a C-terminal 75 kDa truncated protein (WB) [60]. |
Cardiac specific Zebrafish JUPWT/2057_2058del [60]. - The zebrafish mutated myocytes showed significant reduction of INa and IK1 current densities. EM showed disruption of cell–cell contact. (GAL4/UAS transactivation system was used to induce cardiac specific expression of the human 2057_2058del variant in zebrafish) [60]. |
Mismatch, but no patients were traced. |
n.r. | (hom) ACM claimed, but no patients were traced |
Abbreviations: altered protein function > variant annotated in red; partial loss of protein = variant annotated in blue; unclear > variant annotated in grey; US (uncertain significance); LP (likely pathogenic); P (pathogenic); aa (amino acids); n.r. (none reported); n.o. (none observed); NMD (nonsense mediated mRNA decay); fs (frameshift); * or PTC (premature termination codon); WB (Western blot); IF (immunofluorescence); EM (electron microscopy); co-IP (co-immunoprecipitation); WT (wildtype); LV (left ventricle); RV (right ventricle); EBS (epidermolysis bullosa simplex); PPK (palmoplantar keratoderma); WH (woolly hair); ACM (arrhythmogenic cardiomyopathy); hom (homozygous > phenotype observed in homozygous carriers); het (heterozygous > phenotype observed in heterozygous carriers).
3.1. JUP Variants Causing PG Reduction
3.2. JUP Variants Causing an Altered PG Protein
3.3. Potential Therapeutic Avenues
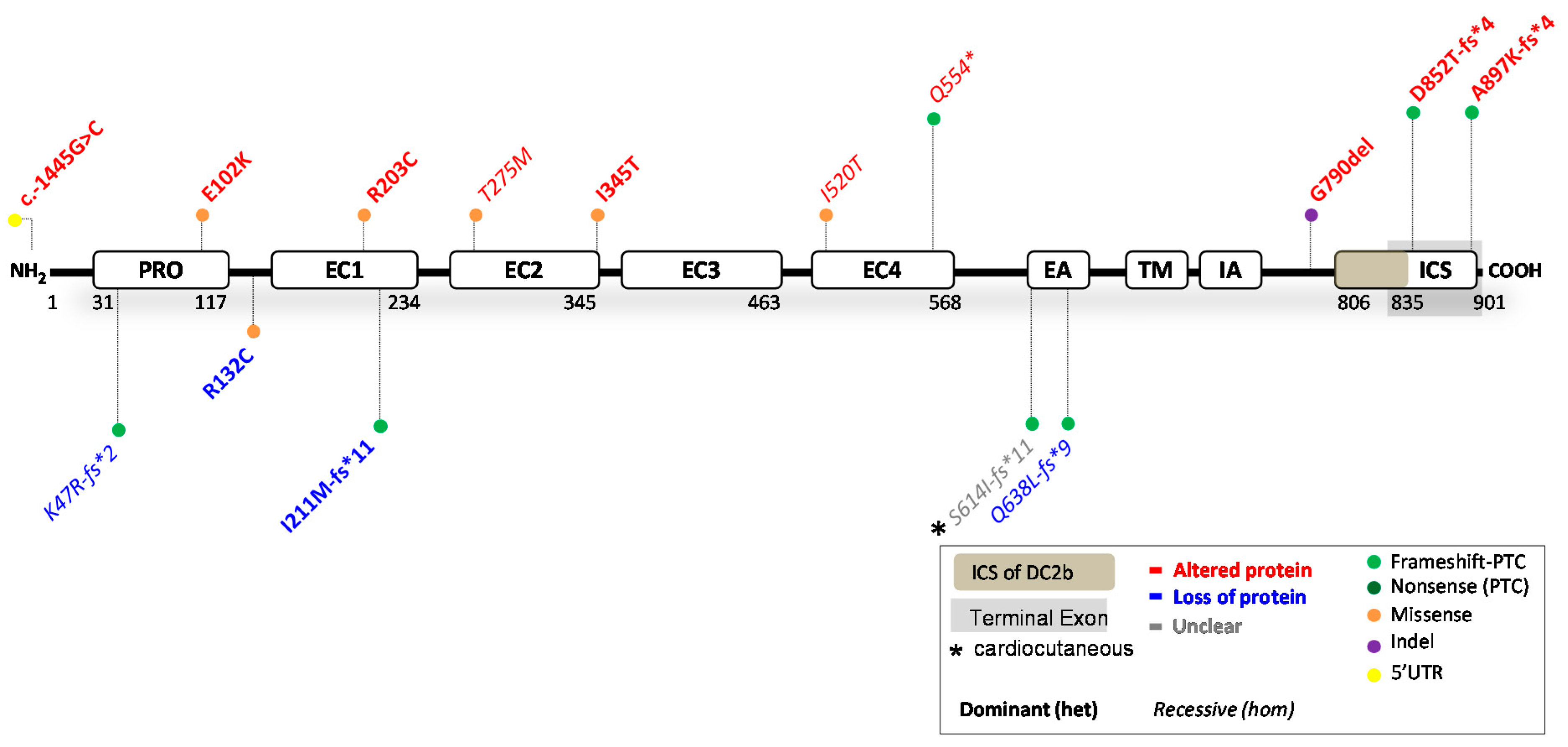
4. Reported DSC2 Variants
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) |
Protein Domain | ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect | Prediction: Functional |
Skin | Heart |
|---|---|---|---|---|---|---|---|---|---|
| c.-1445G > C | NC_000018.10:g.31103416C > G | 5′UTR | B | Not applicable (5′UTR), cannot be predicted |
Altered DC2 function; n.s. -Luciferase assay >a decreased transcriptional activity for HEK cells transfected with the DC2 mutant (c.-1445C) construct [66]. |
Altered transcription factor binding in the presence of the mutant allele. | Mismatch by definition |
n.r. | (het) ACM |
| c.140_147del | p.(Lys47Arg fs*2) | PRO peptide-domain |
LP | >PTC > NMD, no protein | Partial loss of DC2: Patient had reduced levels (>50%) of DC2 in skin biopsy (WB) [67]. |
n.r. -of note: a relative with only DSC2:c.1559T > C (missense) had no phenotype [67]. |
Match | n.r. | (comp.het) NCCM/HCM with DSC2:c.1559T > C |
| c.304G > A | p.(Glu102Lys) | PRO peptide-domain |
LB | >Protein expressed -PolyPhen-2> Benign (0.016) -SIFT> Tolerated -MutPred2> Benign (0.158) |
Altered DC2 function; n.s., but mutant expressed in cells. |
IF shows that the mutant protein localizes in a dotted pattern predominantly in the cytoplasm (COS-1 cells, neonatal rat CM transfection) [68]. | Match | n.r. | (het) ACM |
| c.394C > T | p.(Arg132Cys) | In between PRO peptide and EC1-domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.823) |
Partial loss of DC2; 50% reduced levels of DSC2 mRNA in explanted heart and hiPSC-CMs; reduced DC2 protein in explanted heart (WB) [69] |
-Reduced levels of all desmosomal genes in explanted heart and hiPSC-CMs, reduction of PG at ID in heart; mutant hiPSC-CMs had shortened action potential durations associated with reduced calcium current density and increased potassium current density [69]. -Increased PPARƴ expression and contractile and electric disturbances observed in patient hiPSC-CMs [70] -Zebrafish DSC2WT/c.394C > T [69]. |
Mismatch | n.r. | (het) ACM |
| c.609C > T | p.(Arg203Cys) | EC1 domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.899) |
Altered DC2 function; Complete defect in processing into the mature form [71]. (WB) |
-The mutant protein remains in an unprocessed pro-protein form (COS-1 cells transfection) [71]. -In HL-1 cells, the mutant protein fails to localize at the desmosomes of intercalated disc structures [71]. |
Match | n.r. | (het) ACM |
| c.631-2A > G | p.(Ile211Met fs*11) | EC1 domain |
P | >PTC > NMD, no protein | Partial loss of DC2: Patient heart tissue shows 60% DC2 reduction (WB) [72] -Reduced mRNA DSC2 (only 3% compared to WT) [72] |
-n.r. -Zebrafish KD of DSC2 and DSC2WT/631−2A > G [72]. |
Match | n.o. | (het) ACM |
| c.824C > T | p.(Thr275Met) | EC2 domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Benign (0.563) |
Altered DC2 function; Partial defects in processing into the mature form [71]. (WB) |
-Only a proportion of the partly functional DC2 mutant protein is still incorporated into the desmosomes; affects PG at the intercalated disc (COS-1 cells transfection) [71]. | Match | n.r. | (hom) ACM |
| c.1034T > C | p.(Ile345Thr) | EC2 domain |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.756) -SIFT> NOT tolerated -MutPred2> Benign (0.591) |
Altered DC2 function; n.s., but mutant expressed in cells. |
-In transfected neonatal rat cardiomyocytes and HL-1 cells, the mutant protein localizes in the cytoplasm (IF) [68]. | Match | n.r. | (het) ACM |
| c.1559T > C | p.(Ile520Thr) | EC4 domain |
LB | >Protein expressed -PolyPhen-2> Probably damaging (0.973) -SIFT> NOT tolerated -MutPred2> Benign (0.601) |
Altered DC2 function; Protein is expressed, similar size as wildtype (WB) [67]. Unsure if the protein is really altered, or that it maintains all functions [67]. |
n.r. -of note: a relative with only DSC2:c.1559T > C (missense) had no phenotype [67]. |
Match | n.r. | (comp.het) NCCM/HCM with DSC2:c.140_147del |
| c.1660C > T | p.(Gln554*) | EC4 domain |
P | >PTC > NMD, no protein -MutPredLOF> borderline pathogenic (0.51161) |
Altered DC2 function; Truncated DC2 protein (75 kDa), wildtype is 150 kDa (transfection WB). Less mature protein, more pre-protein than normal [73]. |
-Heart biopsies shows DC2 staining in hom-carriers (protein is expressed); mutant protein localizes only partially at cell membrane and predominantly in cytoplasm (transfection IF WB) [73]. -Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane [74]. |
Mismatch | Mild PPK at pressure points, in one hom- and one het-carrier (possibly secondary to farm work) |
(hom) ACM (LV affected mainly) |
| c.1841del | p.(Ser614Ile fs*11) |
EA domain |
P | >PTC > NMD, no protein | Unclear?; Truncated isoforms expressed (transfection IF, WB), but needs patient cell confirmation. |
Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane [74]. | Unclear | (hom) Mild PPK, WH [75] | (hom) ACM [75] |
| c.1913_1916 del |
p.(Gln638Leu fs*9) |
EA domain |
P | >PTC > NMD, no protein | Partial loss of DC2: Strong DC2 protein reduction in patient heart tissue (<10% left-WB, also IF) [74]. -DSC2 mRNA was decreased in patient heart tissue (qPCR) |
-Patients’ explanted heart shows degradation of sarcomeres and mitochondria; widened intercellular spaces and accumulation of lipid droplets (EM); Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane [74]. | Match | n.o. | (hom) ACM |
| c.2368_2370 del | p.(Gly790del) | In between IA and ICS domains | US | >Protein expressed -MutPred-Indel> NOT pathogenic (0.4309) |
Altered DC2 function: No reduction of DC2 protein levels [76]. |
-Slight LV dysfunction with abnormal calcium release [76]. -Mouse model > [76] Hom-mice (G790del) showed enlarged LV and a decreased fractional shortening. Abnormal intracellular calcium release, but no clear ACM phenotype. Het-mice showed no arrhythmias. |
Match | n.r. | (het) CM |
| c.2553del | p.(Asp852Thr fs*4) |
ICS domain DC2a only |
US | -PTC > terminal exon, not NMD > protein expressed | Altered DC2a function; Truncation of the last 47 aa of the DC2a isoform [77]. |
The mutant protein DC2a lost its ability to bind to PG (HL-1 cells transfection) [77]. | Match | n.r. | (het) ACM |
| c.2687_2688 insGA | p.(Ala897Lys fs*4) | ICS domain DC2a only |
B | -PTC > terminal exon, not NMD > protein expressed | Altered DC2a function; -Premature termination of the protein [78]. -Does not exhibit defects in processing into the mature form [71]. |
-Cytoplasmic localization of the mutant protein (HL-1 cells transfection) [78]. -This mutant protein is processed into its mature form and can be incorporated into desmosomes; impaired binding to DP and PG (COS-1 cells transfection) [71]/ |
Match | n.r. | (het) ACM |
Abbreviations: altered protein function > variant annotated in red; partial loss of protein = variant annotated in blue; unclear > variant annotated in grey; US (uncertain significance); B (benign); LB (likely benign); LP (likely pathogenic); P (pathogenic); n.s. (not specified); aa (amino acids); n.r. (none reported); n.o. (none observed); WT (wildtype); WB (Western blot); IF (immunofluorescence); EM (electron microscopy); NMD (nonsense mediated mRNA decay); fs (frameshift); * or PTC (premature termination codon); hiPSC-CMs (human induced pluripotent stem cell derived cardiomyocytes); KD (knockdown); ID (intercalated disc); PPK (palmoplantar keratoderma); CM (cardiomyopathy); ACM (arrhythmogenic cardiomyopathy); NCCM (non-compaction cardiomyopathy); HCM (hypertrophic cardiomyopathy); LV (left ventricle); hom (homozygous > phenotype observed in homozygous carriers); comp.het (compound heterozygous > phenotype observed in compound heterozygous carriers); het (heterozygous > phenotype observed in heterozygous carriers).
4.1. DSC2 Variants Causing DC2 Reduction
4.2. DSC2 Variants Causing an Altered DC2 Protein
4.3. Potential Therapeutic Avenues
References
- Nekrasova, O.; Green, K.J. Desmosome Assembly and Dynamics. Trends Cell Biol. 2013, 23, 537–546.
- Wan, H.; South, A.P.; Hart, I.R. Increased Keratinocyte Proliferation Initiated through Downregulation of Desmoplakin by RNA Interference. Exp. Cell Res. 2007, 313, 2336–2344.
- Bryson, W.G.; Harland, D.P.; Caldwell, J.P.; Vernon, J.A.; Walls, R.J.; Woods, J.L.; Nagase, S.; Itou, T.; Koike, K. Cortical Cell Types and Intermediate Filament Arrangements Correlate with Fiber Curvature in Japanese Human Hair. J. Struct. Biol. 2009, 166, 46–58.
- Thibaut, S.; Gaillard, O.; Bouhanna, P.; Cannell, D.W.; Bernard, B.A. Human Hair Shape Is Programmed from the Bulb. Br. J. Dermatol. 2005, 152, 632–638.
- Thibaut, S.; Barbarat, P.; Leroy, F.; Bernard, B.A. Human Hair Keratin Network and Curvature. Int. J. Dermatol. 2007, 46, 7–10.
- Gaul, R.T.; Nolan, D.R.; Ristori, T.; Bouten, C.V.C.; Loerakker, S.; Lally, C. Strain Mediated Enzymatic Degradation of Arterial Tissue: Insights into the Role of the Non-Collagenous Tissue Matrix and Collagen Crimp. Acta Biomater. 2018, 77, 301–310.
- Getsios, S.; Huen, A.C.; Green, K.J. Working out the Strength and Flexibility of Desmosomes. Nat. Rev. Mol. Cell Biol. 2004, 5, 271–281.
- Stappenbeck, T.S.; Lamb, J.A.; Corcoran, C.M.; Green, K.J. Phosphorylation of the Desmoplakin COOH Terminus Negatively Regulates Its Interaction with Keratin Intermediate Filament Networks. J. Biol. Chem. 1994, 269, 29351–29354.
- Yang, Z.; Bowles, N.E.; Scherer, S.E.; Taylor, M.D.; Kearney, D.L.; Ge, S.; Nadvoretskiy, V.V.; DeFreitas, G.; Carabello, B.; Brandon, L.I.; et al. Desmosomal Dysfunction Due to Mutations in Desmoplakin Causes Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circ. Res. 2006, 99, 646–655.
- Rasmussen, T.; Hansen, J.; Nissen, P.; Palmfeldt, J.; Dalager, S.; Jensen, U.; Kim, W.; Heickendorff, L.; Mølgaard, H.; Jensen, H.; et al. Protein Expression Studies of Desmoplakin Mutations in Cardiomyopathy Patients Reveal Different Molecular Disease Mechanisms. Clin. Genet. 2013, 84, 20–30.
- Notari, M.; Hu, Y.; Sutendra, G.; Dedeić, Z.; Lu, M.; Dupays, L.; Yavari, A.; Carr, C.A.; Zhong, S.; Opel, A.; et al. IASPP, a Previously Unidentified Regulator of Desmosomes, Prevents Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)-Induced Sudden Death. Proc. Natl. Acad. Sci. USA 2015, 112, E973–E981.
- Basso, C. Ultrastructural Evidence of Intercalated Disc Remodelling in Arrhythmogenic Right Ventricular Cardiomyopathy: An Electron Microscopy Investigation on Endomyocardial Biopsies. Eur. Heart J. 2006, 27, 1847–1854.
- Vermeer, M.C.S.C.; Andrei, D.; Kramer, D.; Nijenhuis, A.M.; Hoedemaekers, Y.M.; Westers, H.; Jongbloed, J.D.H.; Pas, H.H.; Berg, M.P.; Silljé, H.H.W.; et al. Functional Investigation of Two Simultaneous or Separately Segregating DSP Variants within a Single Family Supports the Theory of a Dose-dependent Disease Severity. Exp. Dermatol. 2022, 31, 970–979.
- Bliley, J.M.; Vermeer, M.C.; Duffy, R.M.; Batalov, I.; Kramer, D.; Tashman, J.W.; Shiwarski, D.J.; Lee, A.; Teplenin, A.S.; Volkers, L.; et al. Dynamic Loading of Human Engineered Heart Tissue Enhances Contractile Function and Drives a Desmosome-Linked Disease Phenotype. Sci. Transl. Med. 2021, 13, eabd1817.
- Lin, X.; Ma, Y.; Cai, Z.; Wang, Q.; Wang, L.; Huo, Z.; Hu, D.; Wang, J.; Xiang, M. Next-Generation Sequencing Identified Novel Desmoplakin Frame-Shift Variant in Patients with Arrhythmogenic Cardiomyopathy. BMC Cardiovasc. Disord. 2020, 20, 74.
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.J. Disease Mutations in Desmoplakin Inhibit Cx43 Membrane Targeting Mediated by Desmoplakin-EB1 Interactions. J. Cell Biol. 2014, 206, 779–797.
- Whittock, N.V.; Ashton, G.H.S.; Dopping-Hepenstal, P.J.C.; Gratian, M.J.; Keane, F.M.; Eady, R.A.J.; McGrath, J.A. Striate Palmoplantar Keratoderma Resulting from Desmoplakin Haploinsufficiency. J. Investig. Dermatol. 1999, 113, 940–946.
- Armstrong, D. Haploinsufficiency of Desmoplakin Causes a Striate Subtype of Palmoplantar Keratoderma. Hum. Mol. Genet. 1999, 8, 143–148, Erratum in Hum. Mol. Genet. 1999, 8, 943.
- Wan, H.; Dopping-Hepenstal, P.J.C.J.C.; Gratian, M.J.J.; Stone, M.G.G.; Zhu, G.; Purkis, P.E.E.; South, A.P.P.; Keane, F.; Armstrong, D.K.B.K.B.; Buxton, R.S.S.; et al. Striate Palmoplantar Keratoderma Arising from Desmoplakin and Desmoglein 1 Mutations Is Associated with Contrasting Perturbations of Desmosomes and the Keratin Filament Network. Br. J. Dermatol. 2004, 150, 878–891.
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient Mutations Linked to Arrhythmogenic Cardiomyopathy Enhance Calpain-Mediated Desmoplakin Degradation. JCI Insight 2019, 4, e128643.
- Al-Jassar, C.; Knowles, T.; Jeeves, M.; Kami, K.; Behr, E.; Bikker, H.; Overduin, M.; Chidgey, M. The Nonlinear Structure of the Desmoplakin Plakin Domain and the Effects of Cardiomyopathy-Linked Mutations. J. Mol. Biol. 2011, 411, 1049–1061.
- Boyden, L.M.; Kam, C.Y.; Hernández-Martín, A.; Zhou, J.; Craiglow, B.G.; Sidbury, R.; Mathes, E.F.; Maguiness, S.M.; Crumrine, D.A.; Williams, M.L.; et al. Dominant de Novo DSP Mutations Cause Erythrokeratodermia-Cardiomyopathy Syndrome. Hum. Mol. Genet. 2016, 25, 348–357.
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2005, 112, 636–642.
- Uzumcu, A.; Norgett, E.E.; Dindar, A.; Uyguner, O.; Nisli, K.; Kayserili, H.; Sahin, S.E.; Dupont, E.; Severs, N.J.; Leigh, I.M.; et al. Loss of Desmoplakin Isoform I Causes Early Onset Cardiomyopathy and Heart Failure in a Naxos-like Syndrome. J. Med. Genet. 2006, 43, e5.
- Impact of the DSP-H1684R Genetic Variant on Ion Channels Activity in IPSC-Derived Cardiomyocytes. Cell. Physiol. Biochem. 2020, 54, 696–706.
- Williams, T.; Machann, W.; Kühler, L.; Hamm, H.; Müller-Höcker, J.; Zimmer, M.; Ertl, G.; Ritter, O.; Beer, M.; Schönberger, J. Novel Desmoplakin Mutation: Juvenile Biventricular Cardiomyopathy with Left Ventricular Non-Compaction and Acantholytic Palmoplantar Keratoderma. Clin. Res. Cardiol. 2011, 100, 1087–1093.
- Navarro-Manchón, J.; Fernández, E.; Igual, B.; Asimaki, A.; Syrris, P.; Osca, J.; Salvador, A.; Zorio, E. Miocardiopatía Arritmogénica Con Afectación Predominante del Ventrículo Izquierdo por una Mutación Nueva «sin Sentido» En Desmoplaquina. Rev. Española Cardiol. 2011, 64, 530–534.
- Jonkman, M.F.; Pasmooij, A.M.G.; Pasmans, S.G.M.A.; van den Berg, M.P.; Ter Horst, H.J.; Timmer, A.; Pas, H.H. Loss of Desmoplakin Tail Causes Lethal Acantholytic Epidermolysis Bullosa. Am. J. Hum. Genet. 2005, 77, 653–660.
- Mohammed, F.; Odintsova, E.; Chidgey, M. Missense Mutations in Desmoplakin Plakin Repeat Domains Have Dramatic Effects on Domain Structure and Function. Int. J. Mol. Sci. 2022, 23, 529.
- Mahoney, M.G.; Sadowski, S.; Brennan, D.; Pikander, P.; Saukko, P.; Wahl, J.; Aho, H.; Heikinheimo, K.; Bruckner-Tuderman, L.; Fertala, A.; et al. Compound Heterozygous Desmoplakin Mutations Result in a Phenotype with a Combination of Myocardial, Skin, Hair, and Enamel Abnormalities. J. Investig. Dermatol. 2010, 130, 968–978.
- Favre, B.; Begré, N.; Marsili, L.; van Tintelen, J.P.; Borradori, L. Desmoplakin Gene Variants and Risk for Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002241.
- Favre, B.; Begré, N.; Borradori, L. A Recessive Mutation in the DSP Gene Linked to Cardiomyopathy, Skin Fragility and Hair Defects Impairs the Binding of Desmoplakin to Epidermal Keratins and the Muscle-Specific Intermediate Filament Desmin. Br. J. Dermatol. 2018, 179, 797–799.
- Norgett, E.E. Recessive Mutation in Desmoplakin Disrupts Desmoplakin-Intermediate Filament Interactions and Causes Dilated Cardiomyopathy, Woolly Hair and Keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766.
- Huen, A.C.; Park, J.K.; Godsel, L.M.; Chen, X.; Bannon, L.J.; Amargo, E.V.; Hudson, T.Y.; Mongiu, A.K.; Leigh, I.M.; Kelsell, D.P.; et al. Intermediate Filament–Membrane Attachments Function Synergistically with Actin-Dependent Contacts to Regulate Intercellular Adhesive Strength. J. Cell Biol. 2002, 159, 1005–1017.
- Puzzi, L.; Borin, D.; Martinelli, V.; Mestroni, L.; Kelsell, D.P.; Sbaizero, O. Cellular Biomechanics Impairment in Keratinocytes Is Associated with a C-Terminal Truncated Desmoplakin: An Atomic Force Microscopy Investigation. Micron 2018, 106, 27–33.
- Martherus, R.; Jain, R.; Takagi, K.; Mendsaikhan, U.; Turdi, S.; Osinska, H.; James, J.F.; Kramer, K.; Purevjav, E.; Towbin, J.A. Accelerated Cardiac Remodeling in Desmoplakin Transgenic Mice in Response to Endurance Exercise Is Associated with Perturbed Wnt/β-Catenin Signaling. Am. J. Physiol.-Heart Circ. Physiol. 2016, 38103, H174–H187.
- Albrecht, L.V.; Zhang, L.; Shabanowitz, J.; Purevjav, E.; Towbin, J.A.; Hunt, D.F.; Green, K.J. GSK3- and PRMT-1-Dependent Modifications of Desmoplakin Control Desmoplakin-Cytoskeleton Dynamics. J. Cell Biol. 2015, 208, 597–612.
- Lapouge, K.; Fontao, L.; Champliaud, M.-F.; Jaunin, F.; Frias, M.A.; Favre, B.; Paulin, D.; Green, K.J.; Borradori, L. New Insights into the Molecular Basis of Desmoplakinand Desmin-Related Cardiomyopathies. J. Cell Sci. 2006, 119, 4974–4985.
- Dehner, C.; Rötzer, V.; Waschke, J.; Spindler, V. A Desmoplakin Point Mutation with Enhanced Keratin Association Ameliorates Pemphigus Vulgaris Autoantibody-Mediated Loss of Cell Cohesion. Am. J. Pathol. 2014, 184, 2528–2536.
- Camors, E.M.; Purevjav, E.; Jefferies, J.L.; Saffitz, J.E.; Gong, N.; Ryan, T.D.; Lucky, A.W.; Taylor, M.D.; Sullivan, L.M.; Mestroni, L.; et al. Early Lethality Due to a Novel Desmoplakin Variant Causing Infantile Epidermolysis Bullosa Simplex with Fragile Skin, Aplasia Cutis Congenita, and Arrhythmogenic Cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, e002800.
- Ian Gallicano, G.; Kouklis, P.; Bauer, C.; Yin, M.; Vasioukhin, V.; Degenstein, L.; Fuchs, E. Desmoplakin Is Required Early in Development for Assembly of Desmosomes and Cytoskeletal Linkage. J. Cell Biol. 1998, 143, 2009–2022.
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological Abnormalities Precede Overt Structural Changes in Arrhythmogenic Right Ventricular Cardiomyopathy Due to Mutations in Desmoplakin-A Combined Murine and Human Study. Eur. Heart J. 2012, 33, 1942–1953.
- Garcia-Gras, E. Suppression of Canonical Wnt/ -Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021.
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise Restores Dysregulated Gene Expression in a Mouse Model of Arrhythmogenic Cardiomyopathy. Cardiovasc. Res. 2019, 116, 1199–1213.
- Smith, E.A.; Fuchs, E. Defining the Interactions between Intermediate Filaments and Desmosomes. J. Cell Biol. 1998, 141, 1229–1241.
- Lechler, T.; Fuchs, E. Desmoplakin: An Unexpected Regulator of Microtubule Organization in the Epidermis. J. Cell Biol. 2007, 176, 147–154.
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of Cardiac Wnt/β-Catenin Signalling in Desmoplakin-Deficient AC8 Zebrafish Models Is Rescuable by Genetic and Pharmacological Intervention. Cardiovasc. Res. 2018, 114, 1082–1097.
- Li, D.; Zhang, W.; Liu, Y.; Haneline, L.S.; Shou, W. Lack of Plakoglobin in Epidermis Leads to Keratoderma. J. Biol. Chem. 2012, 287, 10435–10443.
- Cabral, R.M.; Liu, L.; Hogan, C.; Dopping-Hepenstal, P.J.C.; Winik, B.C.; Asial, R.A.; Dobson, R.; Mein, C.A.; Baselaga, P.A.; Mellerio, J.E.; et al. Homozygous Mutations in the 5′ Region of the JUP Gene Result in Cutaneous Disease but Normal Heart Development in Children. J. Investig. Dermatol. 2010, 130, 1543–1550.
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973.
- Huang, H.; Asimaki, A.; Lo, D.; McKenna, W.; Saffitz, J. Disparate Effects of Different Mutations in Plakoglobin on Cell Mechanical Behavior. Cell Motil. Cytoskelet. 2008, 65, 964–978.
- Vahidnezhad, H.; Youssefian, L.; Faghankhani, M.; Mozafari, N.; Saeidian, A.H.; Niaziorimi, F.; Abdollahimajd, F.; Sotoudeh, S.; Rajabi, F.; Mirsafaei, L.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy in Patients with Biallelic JUP-Associated Skin Fragility. Sci. Rep. 2020, 10, 21622.
- Groeneweg, J.A.; Ummels, A.; Mulder, M.; Bikker, H.; van der Smagt, J.J.; van Mil, A.M.; Homfray, T.; Post, J.G.; Elvan, A.; van der Heijden, J.F.; et al. Functional Assessment of Potential Splice Site Variants in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Heart Rhythm 2014, 11, 2010–2017.
- Pigors, M.; Kiritsi, D.; Krümpelmann, S.; Wagner, N.; He, Y.; Podda, M.; Kohlhase, J.; Hausser, I.; Bruckner-Tuderman, L.; Has, C. Lack of Plakoglobin Leads to Lethal Congenital Epidermolysis Bullosa: A Novel Clinico-Genetic Entity. Hum. Mol. Genet. 2011, 20, 1811–1819.
- Liu, L.; Chen, C.; Li, Y.; Yu, R. Whole-Exome Sequencing Identified a De Novo Mutation of Junction Plakoglobin (p.R577C) in a Chinese Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. Biomed. Res. Int. 2019, 2019, 9103860.
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a Deletion in Plakoglobin in Arrhythmogenic Right Ventricular Cardiomyopathy with Palmoplantar Keratoderma and Woolly Hair (Naxos Disease). Lancet 2000, 355, 2119–2124.
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of Myocyte Gap Junctions in Arrhythmogenic Right Ventricular Cardiomyopathy Due to a Deletion in Plakoglobin (Naxos Disease). Heart Rhythm 2004, 1, 3–11.
- Zhang, Z.; Stroud, M.J.; Zhang, J.; Fang, X.; Ouyang, K.; Kimura, K.; Mu, Y.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; et al. Normalization of Naxos Plakoglobin Levels Restores Cardiac Function in Mice. J. Clin. Investig. 2015, 125, 1708–1712.
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac Abnormalities in Familial Palmoplantar Keratosis. Heart 1986, 56, 321–326.
- Asimaki, A.; Kapoor, S.; Plovie, E.; Karin Arndt, A.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74.
- Bierkamp, C.; McLaughlin, K.J.; Schwarz, H.; Huber, O.; Kemler, R. Embryonic Heart and Skin Defects in Mice Lacking Plakoglobin. Dev. Biol. 1996, 180, 780–785.
- Ruiz, P.; Brinkmann, V.; Ledermann, B.; Behrend, M.; Grund, C.; Thalhammer, C.; Vogel, F.; Birchmeier, C.; Günthert, U.; Franke, W.W.; et al. Targeted Mutation of Plakoglobin in Mice Reveals Essential Functions of Desmosomes in the Embryonic Heart. J. Cell Biol. 1996, 135, 215–225.
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.-F.; Haneline, L.S.; Field, L.J.; et al. Restrictive Loss of Plakoglobin in Cardiomyocytes Leads to Arrhythmogenic Cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596.
- Swope, D.; Li, J.; Muller, E.J.; Radice, G.L. Analysis of a Jup Hypomorphic Allele Reveals a Critical Threshold for Postnatal Viability. Genesis 2012, 50, 717–727.
- Kowalczyk, A.P.; Borgwardt, J.E.; Green, K.J. Analysis of Desmosomal Cadherin–Adhesive Function and Stoichiometry of Desmosomal Cadherin-Plakoglobin Complexes. J. Investig. Dermatol. 1996, 107, 293–300.
- Christensen, A.H.; Schmitz, B.; Andersen, C.B.; Bundgaard, H.; Brand, S.-M.; Svendsen, J.H. Functional Promoter Variant in Desmocollin-2 Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2016, 9, 384–387.
- Lin, Y.; Huang, J.; Zhu, Z.; Zhang, Z.; Xian, J.; Yang, Z.; Qin, T.; Chen, L.; Huang, J.; Huang, Y.; et al. Overlap Phenotypes of the Left Ventricular Noncompaction and Hypertrophic Cardiomyopathy with Complex Arrhythmias and Heart Failure Induced by the Novel Truncated DSC2 Mutation. Orphanet J. Rare Dis. 2021, 16, 496.
- Beffagna, G.; De Bortoli, M.; Nava, A.; Salamon, M.; Lorenzon, A.; Zaccolo, M.; Mancuso, L.; Sigalotti, L.; Bauce, B.; Occhi, G.; et al. Missense Mutations in Desmocollin-2 N-Terminus, Associated with Arrhythmogenic Right Ventricular Cardiomyopathy, Affect Intracellular Localization of Desmocollin-2 in Vitro. BMC Med. Genet. 2007, 8, 65.
- Moreau, A.; Reisqs, J.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessière, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 Arrhythmogenic Cardiomyopathy Electrical Instability: From Ion Channels to ECG and Tailored Drug Therapy. Clin. Transl. Med. 2021, 11, e319.
- Reisqs, J.; Moreau, A.; Charrabi, A.; Sleiman, Y.; Meli, A.C.; Millat, G.; Briand, V.; Beauverger, P.; Richard, S.; Chevalier, P. The PPARγ Pathway Determines Electrophysiological Remodelling and Arrhythmia Risks in DSC2 Arrhythmogenic Cardiomyopathy. Clin. Transl. Med. 2022, 12, e748.
- Gehmlich, K.; Syrris, P.; Peskett, E.; Evans, A.; Ehler, E.; Asimaki, A.; Anastasakis, A.; Tsatsopoulou, A.; Vouliotis, A.-I.; Stefanadis, C.; et al. Mechanistic Insights into Arrhythmogenic Right Ventricular Cardiomyopathy Caused by Desmocollin-2 Mutations. Cardiovasc. Res. 2011, 90, 77–87.
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant Desmocollin-2 Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088.
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous Founder Mutation in Desmocollin-2 (DSC2) Causes Arrhythmogenic Cardiomyopathy in the Hutterite Population. Circ. Cardiovasc. Genet. 2013, 6, 327–336.
- Brodehl, A.; Weiss, J.; Debus, J.D.; Stanasiuk, C.; Klauke, B.; Deutsch, M.A.; Fox, H.; Bax, J.; Ebbinghaus, H.; Gärtner, A.; et al. A Homozygous DSC2 Deletion Associated with Arrhythmogenic Cardiomyopathy Is Caused by Uniparental Isodisomy. J. Mol. Cell. Cardiol. 2020, 141, 17–29.
- Simpson, M.A.; Mansour, S.; Ahnood, D.; Kalidas, K.; Patton, M.A.; McKenna, W.J.; Behr, E.R.; Crosby, A.H. Homozygous Mutation of Desmocollin-2 in Arrhythmogenic Right Ventricular Cardiomyopathy with Mild Palmoplantar Keratoderma and Woolly Hair. Cardiology 2009, 113, 28–34.
- Hamada, Y.; Yamamoto, T.; Nakamura, Y.; Sufu-Shimizu, Y.; Nanno, T.; Fukuda, M.; Ono, M.; Oda, T.; Okuda, S.; Ueyama, T.; et al. G790del Mutation in DSC2 Alone Is Insufficient to Develop the Pathogenesis of ARVC in a Mouse Model. Biochem. Biophys. Rep. 2020, 21, 100711.
- Gehmlich, K.; Lambiase, P.D.; Asimaki, A.; Ciaccio, E.J.; Ehler, E.; Syrris, P.; Saffitz, J.E.; McKenna, W.J. A Novel Desmocollin-2 Mutation Reveals Insights into the Molecular Link between Desmosomes and Gap Junctions. Heart Rhythm 2011, 8, 711–718.
- De Bortoli, M.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Smaniotto, G.; Rigato, I.; Calore, M.; Li Mura, I.E.A.; Basso, C.; Thiene, G.; et al. The p.A897KfsX4 Frameshift Variation in Desmocollin-2 Is Not a Causative Mutation in Arrhythmogenic Right Ventricular Cardiomyopathy. Eur. J. Hum. Genet. 2010, 18, 776–782.
- Rimpler, U. Funktionelle Charakterisierung von Desmocollin 2 Während Der Embryonalentwicklung und im Adulten Herzen in Der Maus. Ph.D. Thesis, Humboldt-Universität zu Berlin, Berlin, Germany, 2014.
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019.

