Desmosomes are mirroring, transmembrane protein chains that connect the intermediate filament networks of neighbouring cells. Each chain continuously (dis)assembles due to the turnover of five desmosomal protein types: desmoplakin, plakoglobin, plakophilins, desmocollins and desmogleins. The expression of two genes is critical to the formation of all desmosomes: namely DSP, encoding two differently spliced desmoplakin proteins (DPI and DPII) and JUP, encoding plakoglobin (PG). Meanwhile, plakophilins, desmocollins and desmogleins are expressed in a tissue-specific manner and are therefore encoded by multiple genes.
- cardiocutaneous syndromes
- genotype–phenotype correlation
- functional analysis of genetic variants
1. Introduction
2. Reported DSP Variants
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) | Protein Domain |
ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect | Prediction: Functional | Skin | Prediction: FunctionalHeart | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Skin | Heart | ||||||||||
| c.88G > A | p.(Val30Met) | N-terminus | B | >Protein expressed -PolyPhen-2> Benign (0.000) -SIFT> NOT tolerated -MutPred2> | |||||||
| c.71C > A | p.(Glu2_Met43del) not p.(Ser24*) as predicted |
Head domain Benign (0.092) |
Altered DP function; |
LP | >PTC > NMD, no protein -MutPred-LOF> too short sequence for predictionMutant DP protein expressed, normal size and amount (WB) [9][10 |
Altered PG function; Truncated N-terminal protein (lacking the first 42 aa, translation re-initiation Met43) with reduced expression in the skin (WB) [49]][11]. |
. -Similar JUP mRNA levels as in control [49].Binding to PG abolished (Co-IP); DP localization in cytoplasm (transfection) [9]; DP normal in myocardial and epidermal tissue. Exhibit weaker binding to iASPP (transfection) [11]. Mouse DSPWT/88G > A |
Reduced PG expression (IF; WB) and disrupted distribution of DP and DG1 (IF) [49][9]. | .Match | (het) PPK; (het) WH2 | (het) ACM |
| Mismatch | (hom)EBS; (hom)PPK; (hom)WH | n.o. | c.269A > G | p.(Gln90Arg) | N-terminus | B | >Protein expressed -PolyPhen-2> Probably damaging (0.967) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.757) |
Altered DP function; Mutant DP protein expressed, normal size and amount (WB) [9][11]. |
|||
| c.116_118dup | p.(Ser39dup) | Head domain | Binding to PG abolished (Co-IP); DP localization to cytoplasm (transfection) | US | >Protein expressed -MutPred-Indel> Pathogenic (0.76492) |
Altered PG function: Mutant PG protein size similar as WT (82 kDa; WB) [50][51]. See comment on biological effect [51].[9]. Mouse DSP |
-Patient heart displayed a decrease in signal of DP, PG and Cx43 (IF ) [50] - Transfection of HEK293 with mutant construct showed increased size of PG (90 kDa) due to ubiquitin binding (WB); cytoplasmic localization of mutant-PG (IF); higher proliferation and lower apoptosis; fewer and smaller desmosomes in mutant PG cells (EM ) [WT/269A > G 50][9] |
Match | -Additional binding properties of mutant PG to TAIP-2 and HRC-BP (Co-IP and yeast-two-hybrid) [50] n.s. |
- Diminished cell stiffness, but not cell adhesion [51].(het) ACM | |
| Match | n.o. | (het) ACM | c.273 + 5G > A | ||||||||
| c.201delMultiple splice products |
p.(Ser68Ala fs*92)Intron splice site (N-terminus) |
Head US |
domain-Human Splicing Finder> Broken WT donor site -MaxEntScan> Alteration of WT donor site, probably affecting splicing |
P > Altered splicing, out of frame> PTC > NMD |
>PTC > NMD, no protein | Loss of PG?: Highly reduced levels of JUP mRNA (normal splicing), no WB performed [52] |
-Absence of PG protein staining in skin biopsies (IF); small desmosomes and wide intercellular spaces (EM) [52]. | Match | (hom)EBS; (hom)PPK; (hom) alopecia |
(hom) ACM | |
| c.469-8_469 -1del |
p.(Val157_Lys161del) | Armadillo Domain 1 |
LP | >Protein expressed -MutPred-Indel> Pathogenic (0.78939) |
Unclear; 15 nucleotides shorter cDNA fragments when compared to controls, no WB performed [53]. |
Cryptic splice acceptor site activation in exon 4 [53]. | Unclear | n.r. | (het) ACM | ||
| c.1615C > T | p.(Gln539*) | Armadillo Domain 10 |
P | >PTC > NMD, no protein - MutPred-LOF> Borderline pathogenic (0.62246) |
Loss of PG; No truncated, or full-length PG protein detected in patient’s skin extracts (WB) [54]. -Apart from the strong reduction of JUP (90% reduction), DSP and DSG1 mRNAs were also markedly decreased [54]. |
-Complete loss of PG protein in the patient’s skin (WB and IF, both with N-terminal and C-terminal antibody); No skin barrier formation; significant reduction of DP and DG3 in patient skin (IF). Only few, abnormal desmosomes were formed [54]. - Strong reduction PG in the myocardium [54]. |
Match | (hom) lethal EBS |
n.o. at young age | ||
| c.1729C > T | p.(Arg577Cys) | Armadillo Domain 10 |
LP | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> Tolerated -MutPred2> Pathogenic (0.720) |
Altered PG function: Mutant PG had similar size as WT and was not reduced on blot according to the study [55]. |
-On WB, DG2 and Cx43 protein levels were reduced in mutant expression cells, and desmosomal junctions were destabilized (transfection studies) [55]. | Match | n.r. | (het) ACM | ||
| c.2038_2039del | p.(Trp680Gly fs*11) |
Tail domain |
P | >PTC > NMD, no protein | Altered PG function; C-terminal truncated PG protein is abundantly expressed (56 aa missing) (WB of biopsied LV and RV of multiple patients) [51][56][57]. |
-Reduced Cx43 and PG in patient ventricles and absence of phosphorylated Cx43 (IF; WB). A decreased number of gap junctions in patient’s myocardium (EM) [57]. - Diminishes cell adhesion, but not the cell compliance [51]. -Mouse JUP knockin c.2038_2039del [58]. -The mouse data contradict the human data and suggest that mutant mRNA is broken down by NMD in mice, and not much protein is produced (WB does show truncated protein). The authors reason that although the deletion is located in exon 11, the PTC is located in the terminal exon (exon 12) > homozygous mice die at postnatal day 1, while cardiac development went normal, mice had severe skin fragility. -Fusion of the last 5 exons in mice, produced the truncated protein fully, did not cause lethality; however, mice did not develop cardiac dysfunction at 11 months of age. |
Mismatch Mismatch human: mice |
(hom)PPK; (hom)WH [59] | (hom) ACM [59] | ||
| c.2057_2058del | p.(Met686Asn fs*5) |
Tail domain |
n.a. | >PTC > NMD, no protein | Altered PG function; Transfected myocytes showed a C-terminal 75 kDa truncated protein (WB) [60]. |
Cardiac specific Zebrafish JUPWT/2057_2058del [60]. - The zebrafish mutated myocytes showed significant reduction of INa and IK1 current densities. EM showed disruption of cell–cell contact. (GAL4/UAS transactivation system was used to induce cardiac specific expression of the human 2057_2058del variant in zebrafish) [60]. |
Mismatch, but no patients were traced. |
n.r. | (hom) ACM claimed, but no patients were traced |
Variants Causing PG Reduction
Abbreviations: altered protein function > variant annotated in red; partial loss of protein = variant annotated in blue; unclear > variant annotated in grey; US (uncertain significance); LP (likely pathogenic); P (pathogenic); aa (amino acids); n.r. (none reported); n.o. (none observed); NMD (nonsense mediated mRNA decay); fs (frameshift); * or PTC (premature termination codon); WB (Western blot); IF (immunofluorescence); EM (electron microscopy); co-IP (co-immunoprecipitation); WT (wildtype); LV (left ventricle); RV (right ventricle); EBS (epidermolysis bullosa simplex); PPK (palmoplantar keratoderma); WH (woolly hair); ACM (arrhythmogenic cardiomyopathy); hom (homozygous > phenotype observed in homozygous carriers); het (heterozygous > phenotype observed in heterozygous carriers).
3.1. JUP Variants Causing PG Reduction
3.2.
JUP
Variants Causing an Altered PG Protein
3.3. Potential Therapeutic Avenues
4. Reported DSC2 Variants
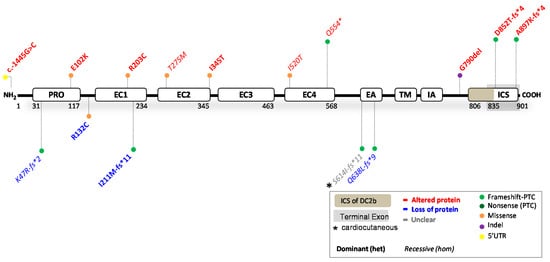
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) |
Protein Domain | ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect | Prediction: Functional |
Skin | Heart | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.-1445G > C | NC_000018.10:g.31103416C > G | 5′UTR | B | Not applicable (5′UTR), cannot be predicted |
Altered DC2 function; n.s. -Luciferase assay >a decreased transcriptional activity for HEK cells transfected with the DC2 mutant (c.-1445C) construct [66]. |
Altered transcription factor binding in the presence of the mutant allele. | Mismatch by definition |
n.r. | (het) ACM | ||||||||||
| c.140_147del | p.(Lys47Arg fs*2) | PRO peptide-domain |
LP | >PTC > NMD, no protein | Partial loss of DC2: Patient had reduced levels (>50%) of DC2 in skin biopsy (WB) [67]. |
n.r. -of note: a relative with only DSC2:c.1559T > C (missense) had no phenotype [67]. |
Match | n.r. | (comp.het) NCCM/HCM with DSC2:c.1559T > C | ||||||||||
| c.304G > A | p.(Glu102Lys) | PRO peptide-domain |
LB | >Protein expressed -PolyPhen-2> Benign (0.016) -SIFT> Tolerated -MutPred2> Benign (0.158)Partial loss of DP: 20% less DP product on WB. No alternatively spliced transcripts discovered in patient-derived cells ([12][13], but did so in in vitro splicing assay (transfection). However, not functional [13]. |
Altered DC2 function; n.s., but mutant expressed in cells.-In combination with c.6687del> Reduced DP protein on blot and staining in explanted heart and hiPSC-CMs and primary KCs [13][14]. -Dislocation of DP after 2D mechanical stretch; resulted in reduced count and density of desmosomes (EM) in dynamic EHTs leading to lower force and stress |
IF shows that the mutant protein localizes in a dotted pattern predominantly in the cytoplasm (COS-1 cells, neonatal rat CM transfection) [68][14]. | .Partial match, normal splicing left. |
(comp.het) PPK; WH |
Matchwith DSP: c.6687delA |
(comp.het) | n.r. ACM/ NCCM :with DSP: c.6687delA |
||||||||
| (het) ACM | c.699G > A | p.(Trp233*) | Plakin-domain | P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.55385) |
Partial loss of DP; Mutant RNA not detected in patient cells. Mutant DP is unstable (transfection-WB) | |||||||||||||
| c.394C > T | p.(Arg132Cys) | In between PRO peptide and EC1-domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.823)[9 |
Partial loss of DC2;]. | 50% reduced levels of DSC2 mRNA in explanted heart and hiPSC-CMs; reduced DC2 protein in explanted heart (WB) [69]Perinuclear aggregates of DP (transfection IF) [9]. |
-Reduced levels of all desmosomal genes in explanted heart and hiPSC-CMs, reduction of PG at ID in heart; mutant hiPSC-CMs had shortened action potential durations associated with reduced calcium current density and increased potassium current density [69]. -Increased PPARƴ expression and contractile and electric disturbances observed in patient hiPSC-CMs [70] -Zebrafish DSC2WT/c.394C > T [69]Partial match, but not with transfection IF/WB |
n.s. | (het) ACM | ||||||||||
| . | Mismatch | n.r. | (het) ACM | c.832del | p.(Ala278Pro fs*39) |
Plakin-domain | P | >PTC > NMD, no protein | |||||||||||
| c.609C > T | Altered DP function; | Truncated DP normally expressed and protein runs at 60 kDa (315 aa) (transfection-WB) | -Leads to truncated DSP mRNA, also indicating that mRNA translation following the truncation was completely impaired. |
c.832del overexpression led to upregulation of PG and downregulation of β-catenin in the nuclei, without affecting their expression in the cytoplasm (transfection) | p.(Arg203Cys) | EC1 domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> [15]. |
Mismatch | n.s. | NOT tolerated (het) ACM |
||||||||
| -MutPred2> | Pathogenic (0.899) | Altered DC2 function; | Complete defect in processing into the mature form [71]. (WB) |
-The mutant protein remains in an unprocessed pro-protein form (COS-1 cells transfection) [71]. -In HL-1 cells, the mutant protein fails to localize at the desmosomes of intercalated disc structures [71]. |
Match | n.r. | (het) ACM | c.861T > G | p.(Asn287Lys) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.997) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.699) |
Altered DP function; Mutant DP expressed [16]. |
Aberrant DP and Cx43 localization (transfection-IF) | |||||
| c.631-2A > G | p.(Ile211Met fs*11) | EC1 domain | [16]. | Match | (hom)PPK; (hom)WH; (hom)EBS | n.o. | |||||||||||||
| P | >PTC > NMD, no protein | Partial loss of DC2: Patient heart tissue shows 60% DC2 reduction (WB) [72] -Reduced mRNA DSC2 (only 3% compared to WT) [72] |
-n.r. -Zebrafish KD of DSC2 and DSC2WT/631−2A > G [72]. |
Match | n.o. | (het) ACM | c.897C > G | p.(Ser299Arg) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.672) |
Altered DP function; Mutant DP expressed [11 | |||||||
| c.824C > T | p.(Thr275Met) | EC2 domain |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Benign (0.563)]. |
Altered DC2 function; Partial defects in processing into the mature form [71Exhibit weaker binding to iASPP = desmosome regulator (transfection) [11]. |
Match | n.s. | (het) ACM | |||||||||||
| ] | . (WB) | -Only a proportion of the partly functional DC2 mutant protein is still incorporated into the desmosomes; affects PG at the intercalated disc (COS-1 cells transfection) | [ | 71 | ]. | Match | n.r. | (hom) ACM | c.939 + 1G > A | p.(Gln331*) | Donor site intron 7 (Plakin-domain) |
P | >PTC > NMD, no protein | ||||||
| c.1034T > C | p.(Ile345Thr) | EC2 domain |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.756) -SIFT> NOT tolerated -MutPred2> Benign (0.591)Partial loss of DP; Absence of detection of mRNA in multiple patient KCs, reported by two studies, suggests efficient NMD [17][ |
Altered DC2 function;18]. Only 20% DPI and 50% DPII is left on WB [19]. | n.s., but mutant expressed in cells.-Major abnormalities in the spinous layer of the epidermis. The intercellular space is widened and KCs contain abnormal cytoplasmic densities [18]. -Small desmosomes and fewer in number; perinuclear keratin distribution 7. - DC3 seems reduced on WB; volume densities of desmosomal proteins seem different from control [19]. |
Match | -In transfected neonatal rat cardiomyocytes and HL-1 cells, the mutant protein localizes in the cytoplasm (IF) [(het) PPK | 68]n.r. | ||||||||||
| . | Match | n.r. | (het) ACM | c.969_974del | p.(Lys324_Glu325del) | Plakin-domain | LP | >Protein expressed -MutPred-Indel> Benign (0.19566) |
Partial loss of DP; Reduced expression of both native DP isoforms in cytoskeletal and cytoplasmic protein fractions (WB) [10], suggesting instable protein> incomplete degradation. | DP expression was significantly reduced in myocardial tissue and epidermal biopsies (IF) [10]. | Mismatch | (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular | ||||||
| c.1559T > C | p.(Ile520Thr) | EC4 domain |
LB | >Protein expressed -PolyPhen-2> Probably damaging (0.973) -SIFT> NOT tolerated -MutPred2> Benign (0.601) |
Altered DC2 function; Protein is expressed, similar size as wildtype (WB) [67]. Unsure if the protein is really altered, or that it maintains all functions [67]. |
n.r. -of note: a relative with only DSC2:c.1559T > C (missense) had no phenotype [67]. |
Match | n.r. | (comp.het) NCCM/HCM with DSC2:c.140_147del | c.1348C > G | p.(Arg451Gly) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Probably | |||||
| c.1660C > T | p.(Gln554*) | EC4 domaindamaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.756) |
Partial loss of DP; 50% reduced DPI&II protein in EHTs (WB) [20]. -mRNA levels of DSP not reduced compared to WT [20]. > instable protein degradation. |
50% reduced DP signal and 70% reduced Cx43 in myocardial tissues (IF); Proteolytic degradation by calpain, leading to DP insufficiency [20]. | Mismatch | n.r. | (het) ACM, bi-ventricular | ||||||||||||
| P | >PTC > NMD, no protein | -MutPredLOF> borderline pathogenic (0.51161) |
Altered DC2 function; Truncated DC2 protein (75 kDa), wildtype is 150 kDa (transfection WB). Less mature protein, more pre-protein than normal [73]. |
-Heart biopsies shows DC2 staining in hom-carriers (protein is expressed); mutant protein localizes only partially at cell membrane and predominantly in cytoplasm (transfection IF WB) [73]. -Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane [74]. |
Mismatch | Mild PPK at pressure points, in one hom- and one het-carrier (possibly secondary to farm work) |
(hom) ACM (LV affected mainly) | c.1372A > T | p.(Asn458Tyr) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Possibly damaging (0.939) -SIFT> Tolerated -MutPred2> Benign (0.323) |
Altered DP function; Mutant DP expressed [16]. |
Altered EB1 binding and Cx43 localization (transfection IF) [16]. | Match | ||||
| c.1841del | p.(Ser614Ile fs*11) |
EA | n.o. | (het) ACM | |||||||||||||||
| domain | P | >PTC > NMD, no protein | Unclear?; Truncated isoforms expressed (transfection IF, WB), but needs patient cell confirmation. |
Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane [74]. | Unclear | (hom) Mild PPK, WH [75] | (hom) ACM [75] | c.1408A > G | p.(Lys470Glu) | Plakin-domain | |||||||||
| c.1913_1916 | US | del |
p.(Gln638Leu >Protein expressed -PolyPhen-2> Benign (0.082) -SIFT> Tolerated -MutPred2> Benign (0.408) |
fs*9)Altered DP function; Conformational alternation, but overall folded structure of DP is remained [21]. Mutant DP expressed (WB) |
EA [16]. |
domain | P | >PTC > NMD, no protein | Partial loss of DC2: Strong DC2 protein reduction in patient heart tissue (<10% left-WB, also IF) [74]. -DSC2Mutant is incorporated into the desmosome [10]. |
Match | n.s./n.o. | (hom) ACM | |||||||
| mRNA was decreased in patient heart tissue (qPCR) | -Patients’ explanted heart shows degradation of sarcomeres and mitochondria; widened intercellular spaces and accumulation of lipid droplets (EM); Transfected cells show that the secreted truncated isoforms are not anchored in the plasma membrane | [ | 74 | ] | . | Match | n.o. | (hom) ACM | c.1598T > C | p.(Ile533Thr) | Plakin-domain | US | |||||||
| c.2368_2370 del | >Protein expressed | -PolyPhen-2> Probably damaging (0.998) |
p.(Gly790del)-SIFT> NOT tolerated -MutPred2> Benign (0.442) |
Altered DP function; Mutant DP expressed (WB) [16]. |
Altered EB1 binding and Cx43 localization (transfection IF) [16]. | Match | n.o. | (het) ACM | |||||||||||
| In between IA and ICS domains | US | >Protein expressed -MutPred-Indel> NOT pathogenic (0.4309) |
Altered DC2 function: No reduction of DC2 protein levels [76]. |
-Slight LV dysfunction with abnormal calcium release [76]. -Mouse model > [76] Hom-mice (G790del) showed enlarged LV and a decreased fractional shortening. Abnormal intracellular calcium release, but no clear ACM phenotype. Het-mice showed no arrhythmias. |
Match | n.r. | (het) CM | c.1696G > A | p.(Ala566Thr) | Plakin-domain | |||||||||
| c.2553del | p.(Asp852Thr US |
fs*4) | ICS domain DC2a only>Protein expressed -PolyPhen-2> Benign (0.007) |
US -SIFT> Tolerated -MutPred2> Benign (0.153) |
Altered DP function; Mutant DP expressed (WB) [16]. |
Mutant is incorporated into the desmosome [ | -PTC > terminal exon, not NMD > protein expressed | Altered DC2a function; Truncation of the last 47 aa of the DC2a isoform [77].10]. |
The mutant protein DC2a lost its ability to bind to PG (HL-1 cells transfection) Match | n.o. | (het) ACM | ||||||||
| [ | 77 | ] | . | Match | n.r. | (het) ACM | c.1853A > C | p.(His618Pro) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Possibly damaging (0.602) -SIFT> NOT tolerated -MutPred2> Benign (0.540) |
Altered DP function; Mutant DP expressed (WB) [22]. |
Mutant localizes to membrane, affected Cx43 localization (transfection studies/skin biopsies). Desmosome aggregation [22]. | Match | (het) PPK; (het) WH; (het) EBS | (het) CM | |||
| c.2687_2688 insGA | p.(Ala897Lys fs*4) | ICS domain DC2a only |
B | -PTC > terminal exon, not NMD > protein expressed | Altered DC2a function; -Premature termination of the protein [78]. -Does not exhibit defects in processing into the mature form [71]. |
-Cytoplasmic localization of the mutant protein (HL-1 cells transfection) [78]. -This mutant protein is processed into its mature form and can be incorporated into desmosomes; impaired binding to DP and PG (COS-1 cells transfection) [71]/ |
Match | n.r. | (het) ACM | c.1865T > C | p.(Leu622Pro) | Plakin-domain | LP | >Protein expressed -PolyPhen-2> Probably damaging (0.998) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.828) |
Altered DP function; Mutant DP expressed (WB) [22]. |
Mutant localizes to membrane, affected Cx43 localization (transfection studies/skin biopsies). Desmosome aggregation [22]. | Match | (het) PPK; (het) WH; (het) EBS | (het) CM |
| c.1755dup | p.(His586Thr fs*9) |
Plakin domain |
P | >PTC > NMD, no protein | Altered DP function; Truncated DP protein, (65 kDa) (WB), truncation of ROD and C-terminus [23]. |
n.r. | Mismatch | n.s. | (het) ACM, LV mostly | ||||||||||
| c.2422C > T | p.(Arg808Cys) | Plakin-domain | US | >Protein expressed -PolyPhen-2> Benign (0.047) -SIFT> NOT tolerated -MutPred2> Benign (0.409) |
Unclear; Conformational alteration (transfection), but overall folded structure of DP is remained [21]. Needs further confirmation in patient cells, whether expressed or not. |
n.r. | Unclear | n.s. | (het) ACM | ||||||||||
| c.3799C > T | p.(Arg1267*) | ROD domain (DPI) |
P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.5552) |
Partial loss of DPI; Instable mutant DPI protein (NMD?) [24]. -Highly reduced DSP mRNA expression (NMD?) [24]. -Complete loss of DPI in patient skin, DPII has normal expression as expected (WB) [24]. |
n.r. | Match | (hom)PPK epidermolytic; (hom)WH |
(hom) ACM/ DCM |
||||||||||
| c.3805C > T | p.(Arg1269*) | ROD domain (DPI) |
P | >PTC > NMD, no protein -MutPred-LOF> Borderline pathogenic (0.55487) |
Partial loss of DPI; Broken down by NMD. DPI/DPI-II protein ratios lower in variant carriers compared with WT individuals. -DPI/DPII expression ratio reduced by 28% in mutant cells. 15-fold lower mutant than WT [10]. |
Decreased DP expression in endomyocardial biopsies. DPI deficiency (IF) [10]. | Match | (het) PPK; (het) WH | (het) DCM, bi-ventricular | ||||||||||
| c.5051A > G | p.(His1684Arg) | ROD domain (DPI) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.956) -SIFT> NOT tolerated -MutPred2> Benign (0.256) |
Altered DPI function: No effect on amount or size of DPI protein on WB [25]. DPII should not be affected. |
Affects action potential and duration; multiple ion channel dysfunction in hiPSC-CMs [25]. | Match | n.r. | (het) CM, conduction disease | ||||||||||
| c.5208_5209del | p.(Gly1737Thrfs*7) | ROD domain (DPI) |
P | >PTC > NMD, no protein | Partial loss of DPI/Unclear? Truncated DPI protein predicted to run at similar height as DPII, yet no increase in this band was observed in skin biopsies (WB) [26]. DPII should not be affected, but data are unclear. |
n.r. | Unclear | (hom) PPK acantholytic; (hom)WH; |
(hom) NCCM, bi-ventricular, severe | ||||||||||
| c.5596C > T | p.(Gln1866*) | ROD domain (DPI) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DPI function; Truncated DPI protein (160 kDa) observed in skin biopsies [27]. DPII should not be affected. |
n.r. | Match | n.s. | (het) ACM, LV dilation | ||||||||||
| c.5800C > T | p.(Arg1934*) | ROD domain (DPI) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DPI function; Truncated DPI protein (243 kDa) (WB) [28]. DPII should not be affected. -Aberrant mRNA transcripts. Not NMD. |
Stable expressed DP protein, which is recruited into desmosomes, although more punctate staining was observed (IF) [28]. | Match | (comp.het) (lethal) EBS, PPK and WH, with DSP: c.6091_6092del [28] |
n.r. | ||||||||||
| c.6091_6092del | p.(Leu2031Glyfs*29) | PRD (A domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Truncated DP-I protein (228 kDa) (WB) [28]. Not clear what happens with DPII. -Aberrant mRNA transcripts. Not NMD. |
Stable expressed DP protein, which is recruited in desmosomes, although more punctate staining was observed (IF) [28]. | Match | (comp.het) (lethal) EBS, PPK; and WH, with DSP: c.5800C > T [28] |
n.r. | ||||||||||
| c.6166G > C | p.(Gly2056Arg) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.872) |
Altered DP function; Expressed in insoluble fraction of bacterial cells (transfection WB) [29]. Low expression in HeLa cells. Likely expressed mutant. |
n.r. | Probable match | (hom) PPK | (hom) ACM, LV involvement | ||||||||||
| c.6247C > T | p.(Arg2083Cys) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Benign (0.443) |
Altered DP function; Expressed in soluble fraction of bacterial cells (transfection WB), thus correctly folded [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) LQTS | ||||||||||
| c.6307A > G | p.(Lys2103Glu) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.860) -SIFT> Tolerated -MutPred2> Benign (0.417) |
Altered DP function; Expressed in soluble fraction of bacterial cell transfection, thus correctly folded (WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) DCM | ||||||||||
| c.6310del | p.(Thr2104Glnfs*12) | PRD (A domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Several truncated DP proteins shown on WB, but mutant is predicted to be 238 kDa [30] |
Fibrosis and fat deposition in the heart with reduction in Cx43, disorganized IDs, but staining of DP, PG and DG2 seemed normal; severe reduction of DPI&II on IF ex vivo skin. β-catenin expression was also reduced on IF in skin [30]. | Match | (comp.het) EBS, PPK and WH: with DSP: c.7964C > A |
(comp.het) Bi-ventricular CM: with DSP: c.7964C > A |
||||||||||
| c.6577G > A | p.(Glu2193Lys) | PRD (A domain) |
US | >Protein expressed -PolyPhen-2> Possibly damaging (0.950) -SIFT> Tolerated -MutPred2> Benign (0.346) |
Altered DP function; Expressed in insoluble fraction in bacterial cells (transfection WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | (comp.het) Alopecia PPK, with DSP:c.7567delAAGA |
(comp.het) DCM, with DSP:c.7567delAAGA | ||||||||||
| c.6687del | p.(Arg2229Serfs*32) | PRD (B domain) |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Partial loss of DP: NMD of product (WB, NMD inhibitor exp.), 50% reduced protein levels [13][14]. -mRNA 50% reduced [14]. |
-Reduced DP protein on blot and staining in explanted heart, hiPSC-CMs and primary KCs [13][14]. -Mislocalisation of DP after 2D mechanical stretch; in combination with c.273 + 5G > A resulted in reduced count and density of desmosomes in hiPSC-derived dynamic EHTs leading to lower force and stress [14] -Faster differentiation observed in primary KCs of patients. Mechanical stretch provoked cell-contact defects [13]. |
Mismatch NMD active in terminal exon! |
(het)PPK; (comp.het)WH: with DSP: c.273 + 5G > A |
Lethal ACM/ NCCM (comp.het) ACM/ NCCM (bi-ventricular) (het) |
||||||||||
| c.6885A > T | p.(Gln2295His) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.833) |
Altered DP function; Likely truncated DP protein expressed. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. | Probable match | n.r. | (hom) DCM | ||||||||||
| c.7012G > A | p.(Gly2338Arg) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.910) |
Altered DP function; Insoluble fraction in bacterial cell transfection (WB) [29]. Likely expressed mutant, needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) CM | ||||||||||
| c.7027G > A | p.(Glu2343Lys) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Benign (0.077) -SIFT> Tolerated -MutPred2> Benign (0.386) |
Altered DP function; Likely truncated DP protein expressed. Soluble fraction in bacterial cell transfection, thus correctly folded (WB) [29]. |
Altered binding with vimentin and keratin8/18 (transfection IF) [31]. | Probable match | n.r. | (het) ACM ACM, with PKP2:c.1468C > T |
||||||||||
| c.7096C > T | p.(Arg2366Cys) | PRD (B domain) |
LP | >Protein expressed -PolyPhen-2> Probably damaging (0.980) -SIFT> NOT tolerated -MutPred2> Benign (0.622) |
Altered DP function; Likely truncated DP protein expressed [31]. Soluble fraction of bacterial cell transfection (WB) [29]. High expression in HeLa cells. Needs confirmation in patient cells. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. No binding deficiency with vimentin (IF transfection) [29]. |
Probable match | (hom)EBS; (hom)PPK; (hom)WH | n.r. | ||||||||||
| c.7123G > C | p.(Gly2375Arg) | PRD (B domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.938) |
Altered DP function; Truncated DP protein expressed [32]. Insoluble fraction of bacterial cell transfection (WB) [29]. |
Co-alignment with IFs severely affected. Diffuse cytosolic distributed [32]. Targeting to IFs affected (transfection-IF) [29]. |
Match | (hom)EBS; (hom)PPK; (hom)WH | (hom)ACM | ||||||||||
| c.7534G > T | p.(Asp2512Tyr) | Linkers | US | >Protein expressed -PolyPhen-2> Probably damaging (0.998) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.825) |
Unclear; Likely truncated DP protein expressed. Needs further confirmation in patient cells. |
No binding deficiency with IFs (transfection-IF) [31]. | Unclear | n.o. | n.o. | ||||||||||
| c.7623G > T | p.(Arg2541Ser) | Linkers | US | >Protein expressed -PolyPhen-2> Benign (0.010) -SIFT> NOT tolerated -MutPred2> Benign (0.265) |
Unclear; Likely truncated DP protein expressed. Needs further confirmation in patient cells. |
No binding deficiency with IFs (transfection-IF) [31]. | Unclear | n.r. | (het) ACM | ||||||||||
| c.7623del | p.(Lys2542Serfs*19) | Linkers | LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Severe reduction of both DPI&II (WB), both truncated proteins detected [10]. |
Normal DP immunoreactivity in epidermal and myocardial tissue (IF)/or almost no signal depending on homozygous or heterozygous patient [10]. | Match | (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular |
||||||||||
| c.7780del | p.(Ser2594Profs*9) | Linkers | LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Truncated DP protein, 18 aa downstream of deletion (WB) [33]. |
Partial disruption with intermediate filament binding (IF) [33]; KCs have alteration in morphology, elasticity, adhesion capabilities and viscoelastic properties [34][35]. | Match | (hom)PPK; (hom)WH | (hom) DCM |
||||||||||
| c.7916G > A | p.(Arg2639Gln) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.978) -SIFT > Tolerated -MutPred2> Benign (0.484) |
Altered DP function; Likely truncated DP proteins expressed [31]. Expressed in soluble fraction in bacterial cells (transfection WB) [29]. |
Altered binding with desmin and keratin8/18 (transfection IF) [31]. No binding deficiency with vimentin (IF transfection) [29]. |
Probable match | n.r. | (het) CM; RV dysfunction |
||||||||||
| c.7940G > A | p.(Gly2647Asp) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.980) -SIFT> NOT tolerated -MutPred2> Benign (0.619) |
Altered DP function: Both in insoluble and soluble fraction in bacterial cells (transfection WB) [29]. Likely expressed mutant. |
n.r. | Probable match | n.r. | (het) CM | ||||||||||
| c.7964C > A | p.(Ala2655Asp) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.999) -SIFT> NOT tolerated -MutPred2> Pathogenic (0.754) |
Altered DP function; Likely truncated DP proteins expressed, as full loss of protein is not expected due to recessive inheritance. |
Severe binding deficiency with intermediate filaments (transfection IF) [31]. | Probable match | (hom)EBS; (hom)PPK; (hom)WH | (hom) ACM, bi-ventricular |
||||||||||
| c.8066A > C | p.(Lys2689Thr) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT > Tolerated -MutPred2> Benign (0.625) |
Altered DP function: Expressed in soluble fraction in bacterial cells (transfection WB), thus correctly folded [29]. High expression in HeLa cells. Likely expressed mutant. Needs confirmation in patient cells. |
No binding deficiency with vimentin (transfection IF) [29]. | Probable match | n.r. | (het) ACM | ||||||||||
| c.8275C > A | p.(Arg2759Ser) | PRD (C domain) |
US | >Protein expressed -PolyPhen-2> Probably damaging (1.000) -SIFT > Tolerated -MutPred2> Benign (0.350) |
Altered DP function: Expressed in soluble fraction in bacterial cells (transfection, thus correctly folded WB) [29]. Likely expressed mutant. Needs confirmation in patient cells. |
n.r. | Probable match | n.r. | (het) ACM | ||||||||||
| c.8501G > A | p.(Arg2834His) | C- terminus |
US | >Protein expressed -PolyPhen-2> Probably damaging (0.972) -SIFT> NOT tolerated -MutPred2> Benign (0.189) |
Altered DP function; C-terminally truncated DP protein (WB) [9]. |
Aberrant IF localization; DP localization at cell membrane; affects other junctional proteins [9]; Arg2834His blocked the GSK3β phosphorylation cascade and reduced DP–GSK3β interactions in KCs and in hearts of Arg2834His DP mice [2]. Mouse DSPWT/8501G > A [9][36][37] |
Match | n.s. | (het) ACM | ||||||||||
| Engineered variant | p.(Ser2849Gly) | C- terminus |
n.a. | >Protein expressed -PolyPhen-2> Probably damaging (0.978) -SIFT> NOT tolerated -MutPred2> Benign (0.344) |
Altered DP function; Mutant DP protein detected (WB), normal size. |
Mutant DP exhibits increased anchorage of keratin/desmin [38] filaments and fosters calcium independency [39]. | n.a. | unknown | unknown | ||||||||||
| c.8576_8577del | p.(Ser2859Leufs*6) | C- terminus |
LP | >PTC > terminal exon, NOT NMD > protein expressed | Altered DP function; Highly reduced mutant DP protein detected in insoluble fraction (WB), none in soluble fraction, but normal size (only 2859 + 6 aa, compared to wildtype 2871 aa) |
GSK3β, normally phosphorylates Ser2859Leu, translocated to the soluble fraction of patient extract where its high activity (dephosph). Ser9 was associated with the phosphorylation (Ser33/37-Thr41) and degradation of β-catenin; abolition of β-catenin phosphorylation in the non-soluble fraction was associated with its translocation into CMs nuclei [40]. | Match | (hom and het) EBS | (hom and het) ACM |
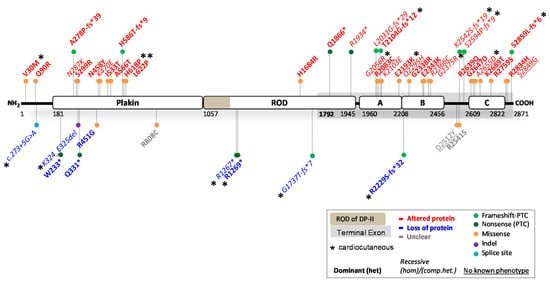
2.1.
DSP
Variants Causing DP Reduction
2.2.
2.1.
DSP
Variants Causing an Altered DP Protein
2.3. Potential Therapeutic Avenues
3. Reported JUP Variants
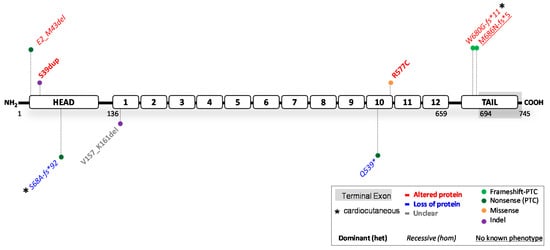
3.1. JUP
| HGVS Nomenclature (DNA) |
HGVS Nomenclature (Protein) |
Protein Domain |
ACMG Class |
In Silico Predictions |
Functional mRNA and Protein Studies |
Biological Effect |
|---|
Abbreviations: altered protein function > variant annotated in red; partial loss of protein = variant annotated in blue; unclear > variant annotated in grey; US (uncertain significance); B (benign); LB (likely benign); LP (likely pathogenic); P (pathogenic); n.s. (not specified); aa (amino acids); n.r. (none reported); n.o. (none observed); WT (wildtype); WB (Western blot); IF (immunofluorescence); EM (electron microscopy); NMD (nonsense mediated mRNA decay); fs (frameshift); * or PTC (premature termination codon); hiPSC-CMs (human induced pluripotent stem cell derived cardiomyocytes); KD (knockdown); ID (intercalated disc); PPK (palmoplantar keratoderma); CM (cardiomyopathy); ACM (arrhythmogenic cardiomyopathy); NCCM (non-compaction cardiomyopathy); HCM (hypertrophic cardiomyopathy); LV (left ventricle); hom (homozygous > phenotype observed in homozygous carriers); comp.het (compound heterozygous > phenotype observed in compound heterozygous carriers); het (heterozygous > phenotype observed in heterozygous carriers).