Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
|
Dermatology
Desmosomes are mirroring, transmembrane protein chains that connect the intermediate filament networks of neighbouring cells. Each chain continuously (dis)assembles due to the turnover of five desmosomal protein types: desmoplakin, plakoglobin, plakophilins, desmocollins and desmogleins. The expression of two genes is critical to the formation of all desmosomes: namely DSP, encoding two differently spliced desmoplakin proteins (DPI and DPII) and JUP, encoding plakoglobin (PG). Meanwhile, plakophilins, desmocollins and desmogleins are expressed in a tissue-specific manner and are therefore encoded by multiple genes.
- cardiocutaneous syndromes
- genotype–phenotype correlation
- functional analysis of genetic variants
1. Introduction
The plakophilin and desmocollin gene family each contain three subtypes (PKP1, PKP2 and PKP3, encoding proteins PP1, PP2 and PP3 and DSC1, DSC2 and DSC3, encoding proteins DC1, DC2 and DC3), while the desmoglein gene family contains four subtypes (DSG1, DSG2, DSG3 and DSG4, encoding proteins DG1, DG2, DG3 and DG4). Keratinocytes of the skin and adnexes express both DP isoforms and PG, in addition to any of the aforementioned PP1-3, DC1-3 and DG1-4 combinations. Each specific composition is in accordance with the differentiation status of keratinocytes in the epidermis [17]. Desmosomal proteins are crucial for epidermal integrity and proper epidermal proliferation and differentiation, and irregularities thereof may cause a skin phenotype [18]. Desmosomal proteins also accommodate hair growth and irregularities thereof. The hair follicle contains keratinocytes in an inner and an outer root sheath. In straight hairs, shafts are straight and homogenous, without clear delimitations, but in coiled hair, these shafts have retro-curvatures [19,20,21]. This curve is achieved via a rotation mechanism of aberrantly proliferating and differentiating cells in the inner root sheath [18,22]. Unlike the skin, the protein composition of desmosomes in cardiac tissue is fixed and consists of DPI, PG, PP2, DC2 and DG2. In the heart, desmosomal anomalies typically disrupt the mechanical continuity of cardiac muscle fibres, which is essential for proper conductance and cardiac muscle contraction. Desmosomes are crucial for the anchorage of cardiomyocytes at the intercalated disc parallel to the direction of strain, where they internally dock the desmin network [16]. Thus far, variants in desmosomal genes DSP, JUP and DSC2 have been associated with a cardiocutaneous phenotype.
2. Reported DSP Variants
DSP encodes for two differently spliced DP proteins: DPI (332 kDa) and a smaller DPII isoform (260 kDa) that contains a shorter rod domain [23]. The latter is created by an alternative donor splice site in exon 23. The N-terminal plakin-domain binds with PPs and PG, while domains B and C at the C-terminal side bind to intermediate filaments (Figure 2). In cardiac muscle, DSP is predominantly spliced into DPI, while the skin contains both isoforms equally. The ClinVar database reported 3290 variants in DSP. Of these, 495 were claimed (likely) pathogenic; 1026 (likely) benign; 161 show conflicting interpretations and 1608 have an unknown significance. Only 48 variants were substantiated by functional evidence, including data from transgenic mouse models (see Figure 2). This indicates that over 98% of all DSP variants were merely predicted by in silico algorithms. For the majority of variants (36/48), the predictions on protein level were correct, while only partially correct in 2/48 variants and incorrect in 5/48 variants. In 5/48 variants, the functional evidence was too elusive to draw conclusions. Moreover, the in silico prediction algorithms frequently contradicted one another, providing little help in assessing the pathogenicity of DSP variants.
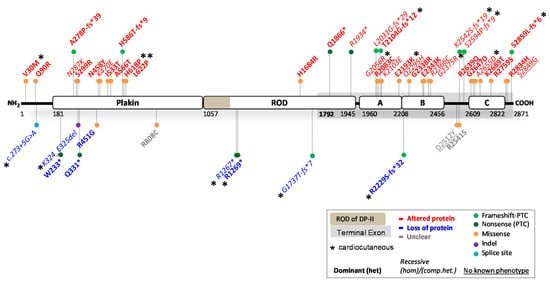
Figure 2. Location of functionally investigated DSP variants.
2.1. DSP Variants Causing DP Reduction
Complete loss of DPI&II is probably incompatible with human life, as it causes early embryonic lethality in mice [55]. More importantly, it has not been functionally observed in human patients. However, several variants that cause protein reduction (<100% native DPI/DPII left) in humans and animal models have been reported [56,57,58]. In total, 9/48 variants resulted in variable degrees of DP reduction and inflicted disease in either a dominant (n = 5) or recessive (n = 4) mode of inheritance. This occurred due to two splice-site variants (c.273 + 5G > A and c.939 + 1G > A), four nonsense-inducing variants (c.699G > A, c.3799C > T, c.3805C > T and c.5208_5209del) located before the terminal exon, and one nonsense-inducing variant in the terminal exon (c.6687del) that was unexpectedly targeted by NMD. Moreover, one missense (c.1348C > G) and one in-frame indel variant (c.969_974del) resulted in DP protein reduction, probably due to instable protein degradation. All of the aforementioned variants affected both isoforms, except for variants c.3799C > T, c.3805C > T and c.5208_5209del. The latter are located in the ROD domain of DPI and therefore only affect DPI, but not DPII. Interpreting the phenotype of patients, DP deficiency (≤50% native DPI) seems to be associated with severe cardiomyopathy, while DP deficiency in the skin (≤50% native DPI and DPII) is mostly associated with PPK and WH. Recessive variants that caused loss of DPI but not DPII, or extreme deficient levels of both DPI and DPII (<20%), were associated with skin fragility [41].
2.1.DSP Variants Causing an Altered DP Protein
The majority of functionally investigated DSP variants (36/48) led to an altered DP protein, due to 23 dominantly and 13 recessively inherited variants. Missense variants were the predominate source for altered DP proteins (n = 27), while the remaining nine variants were due to nonsense or nonsense-inducing variants. Out of the 36 variants, two variants were located near the N-terminus, ten were located in the plakin domain, three in the ROD domain of DPI, six in domain A, five in domain B, two in the linkers, five in domain C and three near the C-terminus. All but one (c.6687del) of the nonsense-inducing variants in the terminal exon of DSP skipped NMD. As expected, the phenotype of patients with an altered DP protein indicated that a recessive mode of inheritance was more severe than a dominant mode of inheritance, mostly due to absence of native protein in the former. Cardiomyopathy was observed in 31/36 of the variant carriers, while it went unobserved or unreported in the others. In the skin, 11/36 variants resulted in PPK (recessive n = 9, dominant n = 2) often with WH (recessive n = 7, dominant n = 2). However, PPK and WH were frequently not observed or not reported in the studies primarily focused on the cardiac phenotype. Furthermore, 10/36 variants caused skin fragility, mostly due to a recessive inheritance (n = 7, dominant n = 3). Variants located in the plakin domain frequently affected the binding efficiency to PG [24,25,26], PPs [59] or intermediate filament anchorage [31,60]. Variants located in domains A, B or C almost always affected the binding affinity to intermediate filaments, especially when causing severe property alterations in domains B and C or loss of these through C-terminal truncation. The functional evidence of the remaining 3/48 variants was inconclusive as to whether it resulted in protein reduction or an altered DP protein.
2.3. Potential Therapeutic Avenues
Given the contradicting results from in silico algorithms and several inconsistencies between the prediction and functional evidence, functional assays of the remaining DSP variants would be strongly encouraged. The nine variants causing DP reduction indicated that the disease severity tends to be dose-dependent in nature, both in the heart as well as in the skin. Hence, strategies to increase native DP protein levels, especially in the heart, would be of benefit to patients. Injections with DSP mRNA in DP-deficient zebrafish have been promising in regaining cardiac function [61]. Besides strategies like RNAi or CRISPR that eliminate protein expression from mutant alleles in patients with altered DP proteins, strategies should simultaneously aim to increase native DP protein levels. For most of the functionally investigated variants, it remains unclear whether they cause a dual organ phenotype, as it is not often assessed or specified.
3. Reported JUP Variants
The JUP gene encodes for the 82 kDa PG protein, also known as ɣ-catenin. PG contains an N-terminal head-domain, 12 armadillo domains and a C-terminal tail-domain (Figure 3). PG belongs to the catenin protein family and is highly homologous to β-catenin, a potent transcription factor of the canonical Wnt/β-catenin signalling pathway. PG is an important desmosomal protein, comprising the outer dense plaque of the desmosome and connecting the transmembrane DG and DC proteins to DP and PP. β-catenin and PG can be substituted for one another, as β-catenin can be incorporated into desmosomes, while PG can also act as a nuclear transcription factor [62]. The ClinVar database has reported 838 variants in JUP. Of these, 30 are claimed (likely) pathogenic; 307 (likely) benign; 70 show conflicting interpretations, and 431 have an unknown significance. Merely eight variants where substantiated by functional evidence, including data from transgenic mouse and zebrafish models (see Figure 3). As for DSP variants, effects of over 98% of all JUP variants were merely predicted by algorithms. The predictions on protein level were correct in 4/8 variants, incorrect in 3/8 variants, while the functional data remained inconclusive in 1/8 variants.
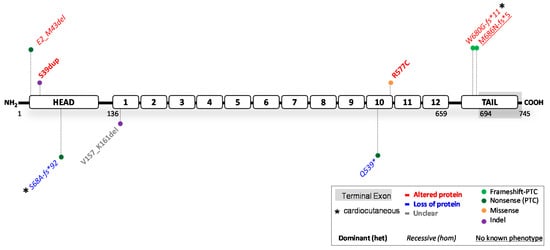
Figure 3. Location of functionally investigated JUP variants.
3.1. JUP Variants Causing PG Reduction
Complete loss of PG induces lethality within embryogenesis in mice due to severe heart defects or immediately post-natal due to severe skin fragility [74,75]. Highly suppressed PG protein levels (<10%) also lead to ACM [76], while 40% protein levels do not induce cardiac dysfunction in mice [77]. This suggests that threshold levels for cardiomyopathy in mice span between 10–40% of native protein. Two human variants (c.201del and c.1615C > T, nonsense) were reported to induce complete PG depletion in homozygous patients [66,68]. These carriers developed severe and sometimes lethal skin fragility, in combination with PPK and alopecia in homozygous c.201del carriers. Cardiomyopathy was not observed in any but one patient with old age [66]. Meanwhile, no other patients with PG reduction (≤50% protein) have been reported. The above results suggest that (near) PG depletion is strongly correlated to skin fragility. However, the dose effect of PG reduction on the development of skin features is unknown and warrants further functional studies. In contrast, while PG depletion induces cardiac lethality during embryogenesis in mice, the limited data currently suggest that the human heart may be protected, perhaps due to functional compensation of β-catenin [62,71]. To accurately assess the cardiac penetrance in patients, more variants predicted to cause reduced or depleted PG levels need to be investigated.
3.2. JUP Variants Causing an Altered PG Protein
Five functionally investigated variants (5/8) resulted in an altered PG protein. These resulted from three recessively inherited nonsense-inducing variants, resulting in either a N-terminal (Glu2_Met43del) or C-terminal (Trp680Glyfs*11 and Met686Asnfs*5) truncated protein. In addition, two dominantly inherited variants, Ser39dup and Arg577Cys, caused ACM and resulted in proteins comparable to the size of native PG. Unlike the C-terminal truncations, the recessive N-terminal truncation induced skin fragility with PPK and WH, but no cardiac dysfunction. The variant causative for this N-terminal truncation c.71C > A, introduces a PTC at Ser24, but translation re-initiation took place at position Met43, which resulted in deletion of the first 42 amino acids. This suggests that any nonsense-inducing variant located between JUP:c.1_126 will likely cause translation re-initiation and a similar phenotype and effect on protein. Opposingly, the two recessive C-terminal truncations correlated with ACM, PPK and WH, but did not induce skin fragility in patients. Moreover, two homozygous PG mouse knockin Trp680Glyfs*11 models were developed, one with and one without fusion of the final five exons. In mice without fusion of exons, this variant resulted in NMD, and only very low levels of C-terminal truncated protein were expressed. These mice died on postnatal day one due to severe skin fragility, induced by depleted PG. Due to their short lifespan, the effect on cardiac function in later stages of development is unclear. In mice with fusion of exons, high levels of C-terminal truncated protein were observed, similarly as in patients. Nonetheless, even at 11 months of age, mice failed to develop cardiac dysfunction [71]. These data suggest that there may be little resemblance between the cardiac function of humans and mice with regard to JUP variants. More variants need to be functionally investigated to draw definitive conclusions.
3.3. Potential Therapeutic Avenues
Currently, too little functional evidence is available to adequately address potential therapeutic strategies that would benefit the cardiac function of patients with disease-causing JUP variants. Meanwhile, strategies to increase native protein in patients with skin fragility seem appropriate for all patients with PG depletion and patients with PG proteins that lack the N-terminus. Whether a skin phenotype is only observed in the case of biallelic JUP variants, as the eight functionally investigated variants now suggest, needs additional functional evidence. Furthermore, almost half of the functionally investigated variants were falsely predicted, which further pressed the need for more functional studies.
4. Reported DSC2 Variants
The DSC2 gene encodes for two transmembrane cadherin isoforms: DC2a (99 kDa) and DC2b (93 kDa). The DC2a isoform contains the complete intracellular segment (ICS), whereas this domain is 53 amino acids shorter in DC2b [78], due to alternative splicing of exon 16 (Figure 4). Both isoforms are first processed into a precursor protein, followed by a mature protein that can be incorporated into desmosomes. Maturely processed DC2 serves as a transmembrane desmosomal protein, important for extracellular cell–cell attachment. The ClinVar database has reported 1209 variants in DSC2. Of these, 102 were claimed (likely) pathogenic; 409 (likely) benign; 85 show conflicting interpretations and 613 have an unknown significance. Notably, merely 15 variants were substantiated by functional evidence, including data from transgenic mouse and zebrafish models (see Figure 4). The same trend seen for DSP and JUP variants, is also observed for DSC2, indicating that over 98% of all variants have not been functionally investigated. The predictions on protein level were correct in 11/15 variants, while incorrect in 3/15 variants and unclear in 1/15 variants.
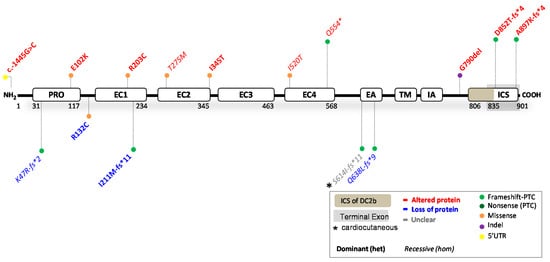
Figure 4. Location of functionally investigated DSC2 variants.
4.1. DSC2 Variants Causing DC2 Reduction
No patients with complete absence of DC2 protein have been reported. Instead, four variants (4/15), located before the terminal exon, resulted in both DC2a and DC2b protein reduction. The recessively (c.1913_1916delAGAA; ≤10% protein left [87]) and dominantly (c.631-2A > G; 40% protein left) inherited nonsense-inducing variants both caused ACM in patients. Compound heterozygosity of out-of-frame indel variant c.140_147delAACTTGT resulted in NCCM and hypertrophy, which is the only functionally investigated DSC2 variant associated with cardiomyopathy other than ACM [80]. The missense variant c.394C > T caused 50% protein reduction via instable protein degradation and caused ACM in a dominant mode of inheritance. Altered electrical properties, a key characteristic of ACM, have been observed in patient hiPSC-CMs containing this missense variant [82]. Moreover, dominantly inherited variants c.394C > T and c.631-2A > G were also investigated in a zebrafish model. The ACM phenotype in both models was rescued by injecting human wildtype but not mutant DSC2 mRNA [82,85]. One of these studies additionally showed that gradual knockdown of DSC2 resulted in dose-dependent cardiac disease severity [85]. This seems to corroborate with the human data, suggesting that cardiomyopathy occurs in situations with ≤50% of native DC2 protein: and the higher the reduction, the more severe the phenotype. Furthermore, in mice, neither complete nor heart-specific knockout of DSC2 resulted in any altered viability or cardiac phenotype [92], which emphasizes differences in disease susceptibility among species. None of the DC2 protein-reducing variants caused a skin phenotype, indicating that near loss of the DC2 protein is well tolerated by the skin. Based on the few investigated variants and contradicting results of animal models, incisive conclusions are still difficult to draw.
4.2. DSC2 Variants Causing an Altered DC2 Protein
Ten variants (10/15) resulted in an altered DC2 protein, which predominantly caused ACM via a dominant (n = 7) or recessive (n = 3) mode of inheritance. Heterozygous variant c.-1445G > C in the 5′UTR affected transcription factor binding mechanisms. Meanwhile, five variants had pronounced effects on the processing of DC2 precursor proteins. For instance, artificial transfection experiments containing missense variants Glu102Lys, Arg203Cys, Thr275Met and Ile345Thr showed punctate cytoplasmic staining, with no or partial ability to be incorporated into desmosomes [84,86,90]. Moreover, nonsense variant Gln554* escaped NMD and resulted in a C-terminal truncated protein, affecting both isoforms [86]. This variant also affected the processing of DC2 precursor proteins, and while a small proportion of maturely processed proteins was incorporated into the desmosome, a larger proportion of precursor proteins remained in the cytoplasm. It is still uncertain whether missense variant Ile520Thr will induce similar alterations that affect the processing of DC2 precursor proteins [80]. Opposingly, nonsense-inducing variants, Asp852Thrfs*4 and Ala897Lysfs*4, only affected isoform DC2a and caused a C-terminal truncated protein. Both were fully processed into a mature protein form, were incorporated into desmosomes, but lost their ability to bind to DP [84] and PG [84,90]. The latter suggests a similar perturbing binding interface for Gln554*. No protein processing information is available on variant Gly790del, other than that it is expressed and translated into a transgenic mouse model. Neither the heterozygous nor homozygous mice showed structural or functional defects in the ventricles or lethal arrhythmias, and only homozygous aged mice showed slight left ventricular dysfunction. This mouse model therefore does not represent the phenotypic severity of the heterozygous Gly790del patients with ACM. In most (7/10) variants, apart from recessive variants Gln554*, Thr275Met and Ile520Thr, ACM was observed in a dominant mode of inheritance. Only recessive inheritance of variant Ser614Ilefs*11 caused PPK and WH, but the functional data were unclear as to whether it causes protein reduction or an altered protein function in patients [87,88]. A skin phenotype was furthermore not observed or went unreported in the other variants.
4.3. Potential Therapeutic Avenues
Whether DSC2 variants can truly cause a cardiocutaneous phenotype remains somewhat elusive, given that only one investigated variant was associated with PPK and WH, and others that do associate with a skin phenotype were not investigated [88]. It seems that extreme deficiency in DC2 is well-compensated for by other desmocollins in the skin (i.e., DC1 and DC3). Nonetheless, more variants should be investigated to draw decisive conclusions. Meanwhile, with the limited functional evidence in mind, patients with DC2 reduction might benefit from native protein-increasing therapeutic strategies. Taken into account, over-administration may be detrimental to humans, as DSC2 overexpression caused severe cardiac dysfunction in mice [93].
This entry is adapted from the peer-reviewed paper 10.3390/ijms231810765
This entry is offline, you can click here to edit this entry!