 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rebecca Katharina Masanetz | -- | 4835 | 2022-10-14 13:58:27 | | | |
| 2 | Jessie Wu | + 19 word(s) | 4854 | 2022-10-18 07:39:38 | | | | |
| 3 | Jessie Wu | Meta information modification | 4854 | 2022-10-18 07:44:36 | | |
Video Upload Options
Inflammatory bowel disease (IBD) is a chronic inflammatory disease comprising two major clinical entities—Crohn’s disease (CD) and ulcerative colitis (UC). IBD incidence remains constantly high in industrialized countries and continuously rises in emerging economies. Importantly, IBD is associated with neuropsychiatric symptoms that strongly worsen IBD disease burden. Mounting evidence indicates that chronic gut inflammation induces a systemic immune response that might cause the CNS manifestation in IBD. In line with this, biologicals targeting inflammatory circuits exerted robust positive effects on depressive symptoms in many autoimmune diseases, and in IBD in particular. Therefore, research in recent years increasingly focused on the characterization of local and systemic immune reactions in IBD, and on entry routes of inflammatory cells and molecules into the CNS. The ultimate aim is to understand how the changes in the neuroimmune landscape impair the function of neurons to cause neuropsychiatric symptoms. In addition, the role of intestinal microbiota in the gut–immune–brain axis in IBD will be discussed.
1. Routes from Peripheral Inflammation to the Central Nervous System in Inflammatory Bowel Disease
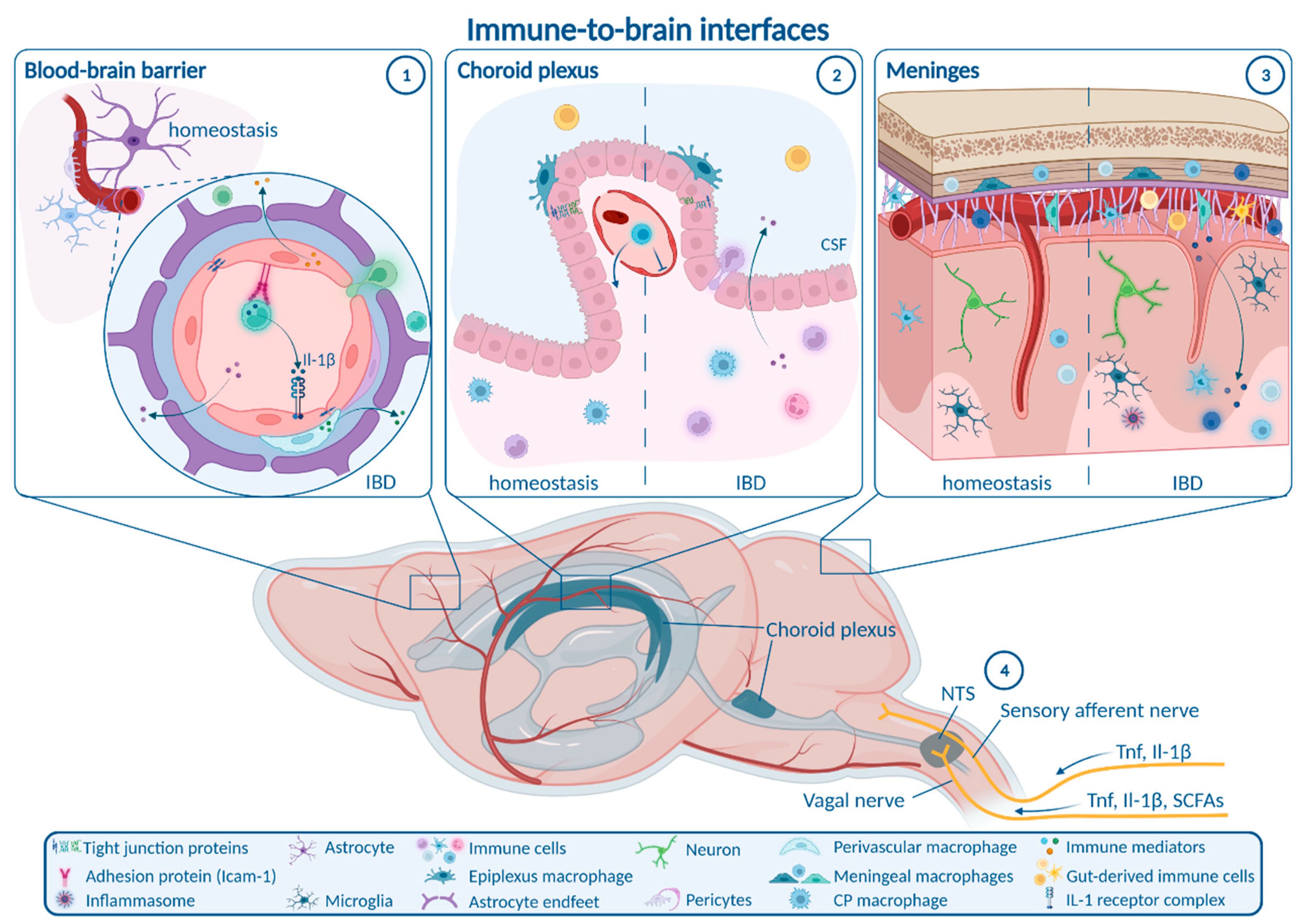
1.1. Enteric, Autonomic and Sensory Nervous System Signaling
1.2. Blood–Brain Barrier
1.3. Choroid Plexus and Blood–CSF Barrier
1.4. Meninges
2. How Neuroinflammation Is Linked to Depression and Anxiety
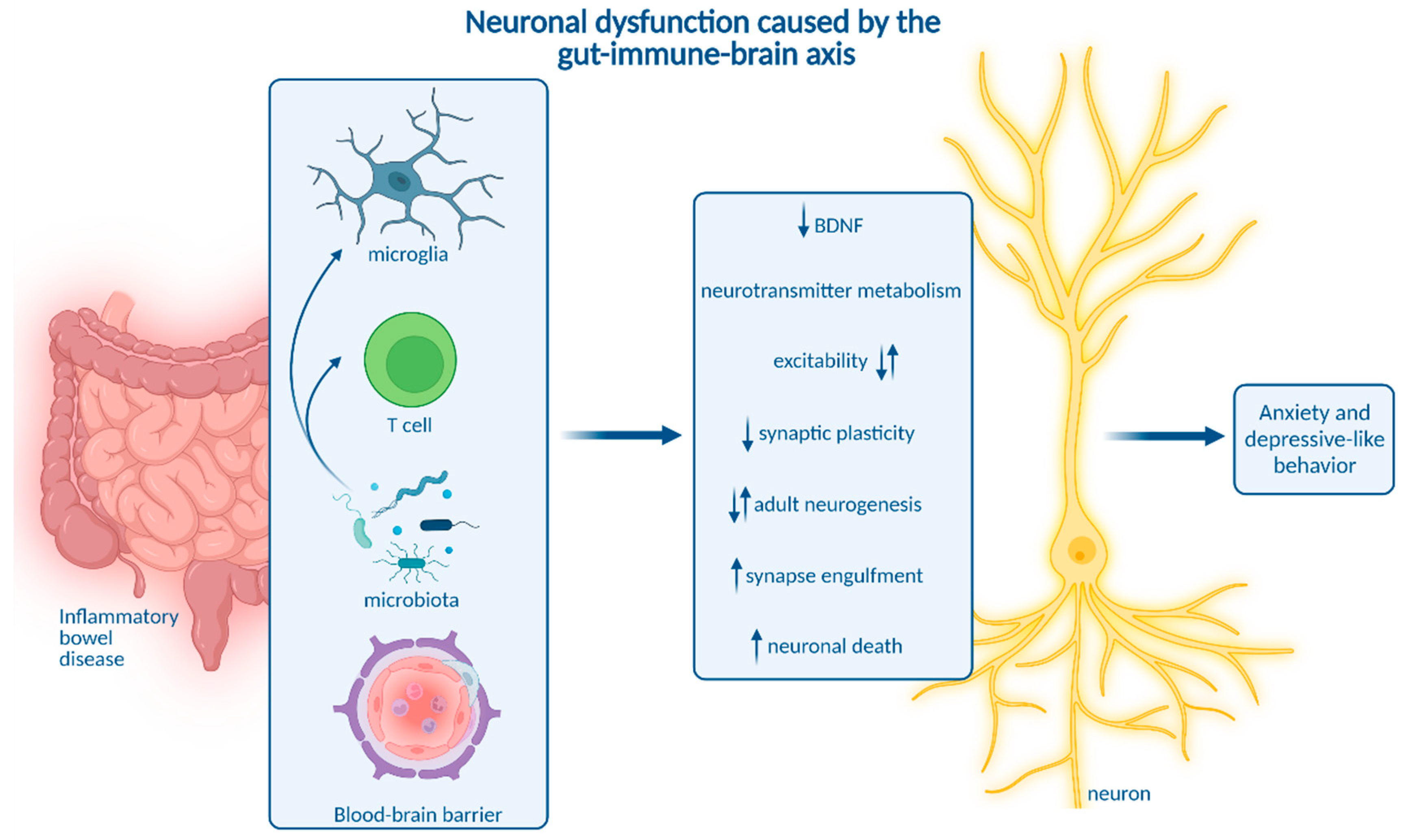
3. Impact of Microbiota on Neuroinflammation and Neuropsychiatric Disease
References
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44.
- Ichiki, T.; Wang, T.; Kennedy, A.; Pool, A.-H.; Ebisu, H.; Anderson, D.J.; Oka, Y. Sensory representation and detection mechanisms of gut osmolality change. Nature 2022, 602, 468–474.
- Goswami, C.; Iwasaki, Y.; Yada, T. Short-chain fatty acids suppress food intake by activating vagal afferent neurons. J. Nutr. Biochem. 2018, 57, 130–135.
- Browning, K.N.; Verheijden, S.; Boeckxstaens, G.E. The Vagus Nerve in Appetite Regulation, Mood, and Intestinal Inflammation. Gastroenterology 2017, 152, 730–744.
- Steinberg, B.E.; Silverman, H.A.; Robbiati, S.; Gunasekaran, M.K.; Tsaava, T.; Battinelli, E.; Stiegler, A.; Bouton, C.E.; Chavan, S.S.; Tracey, K.J.; et al. Cytokine-specific Neurograms in the Sensory Vagus Nerve. Bioelectron. Med. 2016, 3, 7–17.
- Watkins, L.R.; Goehler, L.E.; Relton, J.K.; Tartaglia, N.; Silbert, L.; Martin, D.; Maier, S.F. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: Evidence for vagal mediation of immune-brain communication. Neurosci. Lett. 1995, 183, 27–31.
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic Gastrointestinal Inflammation Induces Anxiety-Like Behavior and Alters Central Nervous System Biochemistry in Mice. Gastroenterology 2010, 139, 2102–2112.e1.
- Sun, P.; Zhou, K.; Wang, S.; Li, P.; Chen, S.; Lin, G.; Zhao, Y.; Wang, T. Involvement of MAPK/NF-κB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS ONE 2013, 8, e69424.
- Sinniger, V.; Pellissier, S.; Fauvelle, F.; Trocmé, C.; Hoffmann, D.; Vercueil, L.; Cracowski, J.L.; David, O.; Bonaz, B. A 12-month pilot study outcomes of vagus nerve stimulation in Crohn’s disease. Neurogastroenterol. Motil. 2020, 32, e13911.
- Ibeakanma, C.; Vanner, S.J. TNI± is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut 2010, 59, 612–621.
- Hess, A.; Roesch, J.; Saake, M.; Sergeeva, M.; Hirschmann, S.; Neumann, H.; Dörfler, A.; Neurath, M.F.; Atreya, R. Functional Brain Imaging Reveals Rapid Blockade of Abdominal Pain Response Upon Anti-TNF Therapy in Crohn’s Disease. Gastroenterology 2015, 149, 864–866.
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131.
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501.
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834.
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12.
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.-H.; Zhao, Y.-Q.; Wu, H.-T.; Fan, M.; Zhu, L.-L. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci. Bull. 2018, 34, 1058–1066.
- Han, Y.; Ding, L.; Cheng, X.; Zhao, M.; Zhao, T.; Guo, L.; Li, X.; Geng, Y.; Fan, M.; Liao, H.; et al. Hypoxia Augments Cerebral Inflammation in a Dextran Sulfate Sodium-Induced Colitis Mouse Model. Front. Cell. Neurosci. 2020, 14, 611764.
- Mitchell, J.; Kim, S.J.; Howe, C.; Lee, S.; Her, J.Y.; Patel, M.; Kim, G.; Lee, J.; Im, E.; Rhee, S.H. Chronic Intestinal Inflammation Suppresses Brain Activity by Inducing Neuroinflammation in Mice. Am. J. Pathol. 2022, 192, 72–86.
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448.
- Zhao, X.; Liang, P.; Liu, J.; Jiang, H.; Fan, X.; Chen, G.; Zhou, C. Elevation of arachidonoylethanolamide levels by activation of the endocannabinoid system protects against colitis and ameliorates remote organ lesions in mice. Exp. Ther. Med. 2017, 14, 5664–5670.
- Hathaway, C.A.; Appleyard, C.B.; Percy, W.H.; Williams, J.L. Experimental colitis increases blood-brain barrier permeability in rabbits. Am. J. Physiol.-Gastrointest. Liver Physiol. 1999, 276, G1174–G1180.
- Natah, S.S.; Mouihate, A.; Pittman, Q.J.; Sharkey, K.A. Disruption of the blood-brain barrier during TNBS colitis. Neurogastroenterol. Motil. 2005, 17, 433–446.
- Barnes, S.E.; Zera, K.A.; Ivison, G.T.; Buckwalter, M.S.; Engleman, E.G. Brain profiling in murine colitis and human epilepsy reveals neutrophils and TNFα as mediators of neuronal hyperexcitability. J. Neuroinflamm. 2021, 18, 199.
- Talley, S.; Valiauga, R.; Anderson, L.; Cannon, A.R.; Choudhry, M.A.; Campbell, E.M. DSS-induced inflammation in the colon drives a proinflammatory signature in the brain that is ameliorated by prophylactic treatment with the S100A9 inhibitor paquinimod. J. Neuroinflamm. 2021, 18, 263.
- Blank, T.; Detje, C.N.; Spieß, A.; Hagemeyer, N.; Brendecke, S.M.; Wolfart, J.; Staszewski, O.; Zöller, T.; Papageorgiou, I.; Schneider, J.; et al. Brain Endothelial- and Epithelial-Specific Interferon Receptor Chain 1 Drives Virus-Induced Sickness Behavior and Cognitive Impairment. Immunity 2016, 44, 901–912.
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000.
- Althubaity, N.; Schubert, J.; Martins, D.; Yousaf, T.; Nettis, M.A.; Mondelli, V.; Pariante, C.; Harrison, N.A.; Bullmore, E.T.; Dima, D.; et al. Choroid plexus enlargement is associated with neuroinflammation and reduction of blood brain barrier permeability in depression. NeuroImage Clin. 2022, 33, 102926.
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus—derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183.
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Antunes, A.R.P.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035.
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93.
- Dempsey, E.; Abautret-Daly, Á.; Docherty, N.G.; Medina, C.; Harkin, A. Persistent central inflammation and region specific cellular activation accompany depression- and anxiety-like behaviours during the resolution phase of experimental colitis. Brain Behav. Immun. 2019, 80, 616–632.
- Filiano, A.J.; Xu, Y.; Tustison, N.; Marsh, R.L.; Baker, W.; Smirnov, I.; Overall, C.C.; Gadani, S.P.; Turner, S.; Weng, Z.; et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 2016, 535, 425–429.
- De Lima, K.A.; Rustenhoven, J.; Da Mesquita, S.; Wall, M.; Salvador, A.F.; Smirnov, I.; Cebinelli, G.M.; Mamuladze, T.; Baker, W.; Papadopoulos, Z.; et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat. Immunol. 2020, 21, 1421–1429.
- Brea, D.; Poon, C.; Benakis, C.; Lubitz, G.; Murphy, M.; Iadecola, C.; Anrather, J. Stroke affects intestinal immune cell trafficking to the central nervous system. Brain Behav. Immun. 2021, 96, 295–302.
- Kivisäkk, P.; Tucky, B.; Wei, T.; Campbell, J.J.; Ransohoff, R.M. Human cerebrospinal fluid contains CD4+ memory T cells expressing gut- or skin-specific trafficking determinants: Relevance for immunotherapy. BMC Immunol. 2006, 7, 14.
- He, X.-F.; Li, L.-L.; Xian, W.-B.; Li, M.-Y.; Zhang, L.-Y.; Xu, J.-H.; Pei, Z.; Zheng, H.-Q.; Hu, X.-Q. Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. J. Neuroinflamm. 2021, 18, 153.
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423.
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609.
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.e24.
- Klawonn, A.M.; Fritz, M.; Castany, S.; Pignatelli, M.; Canal, C.; Similä, F.; Tejeda, H.A.; Levinsson, J.; Jaarola, M.; Jakobsson, J.; et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021, 54, 225–234.e6.
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e9.
- Wohleb, E.S.; Terwilliger, R.; Duman, C.H.; Duman, R.S. Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol. Psychiatry 2018, 83, 38–49.
- Ji, C.; Tang, Y.; Zhang, Y.; Li, C.; Liang, H.; Ding, L.; Xia, X.; Xiong, L.; Qi, X.-R.; Zheng, J.C. Microglial glutaminase 1 deficiency mitigates neuroinflammation associated depression. Brain Behav. Immun. 2022, 99, 231–245.
- Li, W.; Ali, T.; He, K.; Liu, Z.; Shah, F.A.; Ren, Q.; Liu, Y.; Jiang, A.; Li, S. Ibrutinib alleviates LPS-induced neuroinflammation and synaptic defects in a mouse model of depression. Brain Behav. Immun. 2021, 92, 10–24.
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.; Rodrigues, A.L.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain Behav. Immun. 2017, 64, 367–383.
- Dang, R.; Wang, M.; Li, X.; Wang, H.; Liu, L.; Wu, Q.; Zhao, J.; Ji, P.; Zhong, L.; Licinio, J.; et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J. Neuroinflamm. 2022, 19, 41.
- Ali, T.; Hao, Q.; Ullah, N.; Rahman, S.U.; Shah, F.A.; He, K.; Zheng, C.; Li, W.; Murtaza, I.; Li, Y.; et al. Melatonin Act as an Antidepressant via Attenuation of Neuroinflammation by Targeting Sirt1/Nrf2/HO-1 Signaling. Front. Mol. Neurosci. 2020, 13, 96.
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Perez, C.F.; Golden, S.A.; Tamminga, C.; Turecki, G.; et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. USA 2020, 117, 3326–3336.
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760.
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 2018, 23, 1421–1431.
- Zheng, Z.-H.; Tu, J.-L.; Li, X.-H.; Hua, Q.; Liu, W.-Z.; Liu, Y.; Pan, B.-X.; Hu, P.; Zhang, W.-H. Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain Behav. Immun. 2021, 91, 505–518.
- Riazi, K.; Galic, M.A.; Kuzmiski, J.B.; Ho, W.; Sharkey, K.A.; Pittman, Q.J. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 17151–17156.
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4.
- Ghia, J.E.; Li, N.; Wang, H.; Collins, M.; Deng, Y.; El-Sharkawy, R.T.; Côté, F.; Mallet, J.; Khan, W.I. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology 2009, 137, 1649–1660.
- Shajib, M.S.; Baranov, A.; Khan, W.I. Diverse Effects of Gut-Derived Serotonin in Intestinal Inflammation. ACS Chem. Neurosci. 2017, 8, 920–931.
- Kristensen, M.S.; Kjærulff, T.M.; Ersbøll, A.K.; Green, A.; Hallas, J.; Thygesen, L.C. The Influence of Antidepressants on the Disease Course among Patients with Crohn’s Disease and Ulcerative Colitis-A Danish Nationwide Register-Based Cohort Study. Inflamm. Bowel Dis. 2019, 25, 886–893.
- Chen, L.-M.; Bao, C.-H.; Wu, Y.; Liang, S.-H.; Wang, D.; Wu, L.-Y.; Huang, Y.; Liu, H.-R.; Wu, H.-G. Tryptophan-kynurenine metabolism: A link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflamm. 2021, 18, 135.
- Sroor, H.M.; Hassan, A.M.; Zenz, G.; Valadez-Cosmes, P.; Farzi, A.; Holzer, P.; El-Sharif, A.; Gomaa, F.A.-Z.M.; Kargl, J.; Reichmann, F. Experimental colitis reduces microglial cell activation in the mouse brain without affecting microglial cell numbers. Sci. Rep. 2019, 9, 20217.
- Süß, P.; Schlachetzki, J.C.M. Microglia in Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 29–43.
- Crider, A.; Feng, T.; Pandya, C.D.; Davis, T.; Nair, A.; Ahmed, A.O.; Baban, B.; Turecki, G.; Pillai, A. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav. Immun. 2018, 70, 246–256.
- Bolton, J.L.; Short, A.K.; Othy, S.; Kooiker, C.L.; Shao, M.; Gunn, B.G.; Beck, J.; Bai, X.; Law, S.M.; Savage, J.C.; et al. Early stress-induced impaired microglial pruning of excitatory synapses on immature CRH-expressing neurons provokes aberrant adult stress responses. Cell Rep. 2022, 38, 110600.
- Vadodaria, K.C.; Gage, F.H. SnapShot: Adult Hippocampal Neurogenesis. Cell 2014, 156, 1114–1114.e1.
- Jacobs, B.L.; van Praag, H.; Gage, F.H. Adult brain neurogenesis and psychiatry: A novel theory of depression. Mol. Psychiatry 2000, 5, 262–269.
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87.
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.; Enikolopov, G.; Wadiche, L.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495.
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.W.; Lindvall, O. Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. J. Neurosci. 2006, 26, 9703–9712.
- Goshen, I.; Kreisel, T.; Ben-Menachem-Zidon, O.; Licht, T.; Weidenfeld, J.; Ben-Hur, T.; Yirmiya, R. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 2007, 13, 717–728.
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760–1765.
- Korolkova, O.Y.; Myers, J.N.; Pellom, S.T.; Wang, L.; M’Koma, A.E. Characterization of Serum Cytokine Profile in Predominantly Colonic Inflammatory Bowel Disease to Delineate Ulcerative and Crohn’s Colitides. Clin. Med. Insights Gastroenterol. 2015, 8, 29–44.
- Tatsuki, M.; Hatori, R.; Nakazawa, T.; Ishige, T.; Hara, T.; Kagimoto, S.; Tomomasa, T.; Arakawa, H.; Takizawa, T. Serological cytokine signature in paediatric patients with inflammatory bowel disease impacts diagnosis. Sci. Rep. 2020, 10, 14638.
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94.
- Gampierakis, I.-A.; Koutmani, Y.; Semitekolou, M.; Morianos, I.; Polissidis, A.; Katsouda, A.; Charalampopoulos, I.; Xanthou, G.; Gravanis, A.; Karalis, K.P. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Mol. Psychiatry 2021, 26, 1248–1263.
- Salvo, E.; Stokes, P.; Keogh, C.E.; Brust-Mascher, I.; Hennessey, C.; Knotts, T.A.; Sladek, J.A.; Rude, K.M.; Swedek, M.; Rabasa, G.; et al. A murine model of pediatric inflammatory bowel disease causes microbiota-gut-brain axis deficits in adulthood. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G361–G374.
- Zonis, S.; Pechnick, R.N.; Ljubimov, V.A.; Mahgerefteh, M.; Wawrowsky, K.; Michelsen, K.S.; Chesnokova, V. Chronic intestinal inflammation alters hippocampal neurogenesis. J. Neuroinflamm. 2015, 12, 65.
- Nakagawasai, O.; Yamada, K.; Takahashi, K.; Odaira, T.; Sakuma, W.; Ishizawa, D.; Takahashi, N.; Onuma, K.; Hozumi, C.; Nemoto, W.; et al. Liver hydrolysate prevents depressive-like behavior in an animal model of colitis: Involvement of hippocampal neurogenesis via the AMPK/BDNF pathway. Behav. Brain Res. 2020, 390, 112640.
- Takahashi, K.; Kurokawa, K.; Miyagawa, K.; Mochida-Saito, A.; Nemoto, Y.; Iwasa, H.; Nakagawasai, O.; Tadano, T.; Takeda, H.; Tsuji, M. Antidementia effects of Enterococcus faecalis 2001 are associated with enhancement of hippocampal neurogenesis via the ERK-CREB-BDNF pathway in olfactory bulbectomized mice. Physiol. Behav. 2020, 223, 112997.
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23.
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19.
- Barrientos, R.; Sprunger, D.; Campeau, S.; Higgins, E.; Watkins, L.; Rudy, J.; Maier, S. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847–853.
- Zhuang, X.; Zhan, B.; Jia, Y.; Li, C.; Wu, N.; Zhao, M.; Chen, N.; Guo, Y.; Du, Y.; Zhang, Y.; et al. IL-33 in the basolateral amygdala integrates neuroinflammation into anxiogenic circuits via modulating BDNF expression. Brain Behav. Immun. 2022, 102, 98–109.
- Haj-Mirzaian, A.; Amiri, S.; Amini-Khoei, H.; Hosseini, M.-J.; Haj-Mirzaian, A.; Momeny, M.; Rahimi-Balaei, M.; Dehpour, A.R. Anxiety- and Depressive-Like Behaviors are Associated with Altered Hippocampal Energy and Inflammatory Status in a Mouse Model of Crohn’s Disease. Neuroscience 2017, 366, 124–137.
- Heydarpour, P.; Rahimian, R.; Fakhfouri, G.; Khoshkish, S.; Fakhraei, N.; Salehi-Sadaghiani, M.; Wang, H.; Abbasi, A.; Dehpour, A.R.; Ghia, J.E. Behavioral despair associated with a mouse model of Crohn’s disease: Role of nitric oxide pathway. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 131–141.
- Yuan, X.; Chen, B.; Duan, Z.; Xia, Z.; Ding, Y.; Chen, T.; Liu, H.; Wang, B.; Yang, B.; Wang, X.; et al. Depression and anxiety in patients with active ulcerative colitis: Crosstalk of gut microbiota, metabolomics and proteomics. Gut Microbes 2021, 13, 1987779.
- Vicentini, F.A.; Szamosi, J.C.; Rossi, L.; Griffin, L.; Nieves, K.; Bihan, D.; Lewis, I.A.; Pittman, Q.J.; Swain, M.G.; Surette, M.G.; et al. Colitis-associated microbiota drives changes in behaviour in male mice in the absence of inflammation. Brain Behav. Immun. 2022, 102, 266–278.
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632.
- Zhang, Y.; Fan, Q.; Hou, Y.; Zhang, X.; Yin, Z.; Cai, X.; Wei, W.; Wang, J.; He, D.; Wang, G.; et al. Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain Behav. Immun. 2022, 102, 11–22.
- Takahashi, K.; Nakagawasai, O.; Nemoto, W.; Odaira, T.; Sakuma, W.; Onogi, H.; Nishijima, H.; Furihata, R.; Nemoto, Y.; Iwasa, H.; et al. Effect of Enterococcus faecalis 2001 on colitis and depressive-like behavior in dextran sulfate sodium-treated mice: Involvement of the brain–gut axis. J. Neuroinflamm. 2019, 16, 201.
- Emge, J.R.; Huynh, K.; Miller, E.N.; Kaur, M.; Reardon, C.; Barrett, K.E.; Gareau, M.G. Modulation of the microbiota-gut-brain axis by probiotics in a murine model of inflammatory bowel disease. Am. J. Physiol. Liver Physiol. 2016, 310, G989–G998.
- Sankowski, R.; Ahmari, J.; Mezö, C.; de Angelis, A.L.H.; Fuchs, V.; Utermöhlen, O.; Buch, T.; Blank, T.; de Agüero, M.G.; Macpherson, A.J.; et al. Commensal microbiota divergently affect myeloid subsets in the mammalian central nervous system during homeostasis and disease. EMBO J. 2021, 40, e108605.
- Rodríguez-Nogales, A.; Algieri, F.; Garrido-Mesa, J.; Vezza, T.; Utrilla, M.P.; Chueca, N.; García, F.; Rodríguez-Cabezas, M.E.; Gálvez, J. Intestinal anti-inflammatory effect of the probiotic Saccharomyces boulardii in DSS-induced colitis in mice: Impact on microRNAs expression and gut microbiota composition. J. Nutr. Biochem. 2018, 61, 129–139.
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7.
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977.
- Mossad, O.; Batut, B.; Yilmaz, B.; Dokalis, N.; Mezö, C.; Nent, E.; Nabavi, L.S.; Mayer, M.; Maron, F.J.M.; Buescher, J.M.; et al. Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N6-carboxymethyllysine. Nat. Neurosci. 2022, 25, 295–305.
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16.
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728.
- Leonardi, I.; Gao, I.H.; Lin, W.-Y.; Allen, M.; Li, X.V.; Fiers, W.D.; De Celie, M.B.; Putzel, G.G.; Yantiss, R.K.; Johncilla, M.; et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 2022, 185, 831–846.e14.
- Bittel, M.; Reichert, P.; Sarfati, I.; Dressel, A.; Leikam, S.; Uderhardt, S.; Stolzer, I.; Phu, T.A.; Ng, M.; Vu, N.K.; et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J. Extracell. Vesicles 2021, 10, e12159.
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides Species Induce Colitis in Host-Genotype-Specific Fashion in a Mouse Model of Inflammatory Bowel Disease. Cell Host Microbe 2011, 9, 390–403.
- Dicks, L.M.T.; Hurn, D.; Hermanus, D. Gut Bacteria and Neuropsychiatric Disorders. Microorganisms 2021, 9, 2583.
- Chen, J.-J.; Zeng, B.-H.; Li, W.-W.; Zhou, C.-J.; Fan, S.-H.; Cheng, K.; Zeng, L.; Zheng, P.; Fang, L.; Wei, H.; et al. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav. Brain Res. 2017, 322, 34–41.
- Hoban, A.E.; Stilling, R.M.; Moloney, G.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. The microbiome regulates amygdala-dependent fear recall. Mol. Psychiatry 2018, 23, 1134–1144.
- Casado-Bedmar, M.; Viennois, E. MicroRNA and Gut Microbiota: Tiny but Mighty—Novel Insights into Their Cross-talk in Inflammatory Bowel Disease Pathogenesis and Therapeutics. J. Crohn’s Colitis 2021, 16, 992–1005.
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L.; et al. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548.
- Mossad, O.; Nent, E.; Woltemate, S.; Folschweiller, S.; Buescher, J.M.; Schnepf, D.; Erny, D.; Staeheli, P.; Bartos, M.; Szalay, A.; et al. Microbiota-dependent increase in δ-valerobetaine alters neuronal function and is responsible for age-related cognitive decline. Nat. Aging 2021, 1, 1127–1136.
- Vicentini, F.A.; Keenan, C.M.; Wallace, L.E.; Woods, C.; Cavin, J.-B.; Flockton, A.R.; Macklin, W.B.; Belkind-Gerson, J.; Hirota, S.A.; Sharkey, K.A. Intestinal microbiota shapes gut physiology and regulates enteric neurons and glia. Microbiome 2021, 9, 210.

