Inflammatory bowel disease (IBD) is a chronic inflammatory disease comprising two major clinical entities—Crohn’s disease (CD) and ulcerative colitis (UC). IBD incidence remains constantly high in industrialized countries and continuously rises in emerging economies. Importantly, IBD is associated with neuropsychiatric symptoms that strongly worsen IBD disease burden. Mounting evidence indicates that chronic gut inflammation induces a systemic immune response that might cause the CNS manifestation in IBD. In line with this, biologicals targeting inflammatory circuits exerted robust positive effects on depressive symptoms in many autoimmune diseases, and in IBD in particular. Therefore, research in recent years increasingly focused on the characterization of local and systemic immune reactions in IBD, and on entry routes of inflammatory cells and molecules into the CNS. The ultimate aim is to understand how the changes in the neuroimmune landscape impair the function of neurons to cause neuropsychiatric symptoms. In addition, the role of intestinal microbiota in the gut–immune–brain axis in IBD will be discussed.
- inflammatory bowel disease
- neuroinflammation
- gut microbiota
- Crohn's disease
- ulcerative colitis
- systemic inflammation
- depression
- gut-brain axis
1. Routes from Peripheral Inflammation to the Central Nervous System in Inflammatory Bowel S in IBDisease
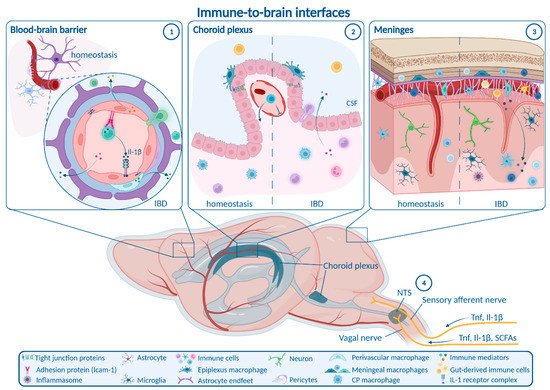
1.1. Enteric, Autonomic and Sensory Nervous System Signaling
1.1. Enteric, Autonomic and Sensory Nervous System Signaling
1.2. Blood–Brain Barrier
1.3. Choroid Plexus and Blood–CSF Barrier
1.4. Meninges
2. How Neuroinflammation Is Linked to Depression and Anxiety
CNS immune activation is a well-known phenomenon in depression and anxiety. Recent findings indicate that immune processes represent a key pathogenic driver rather than a pure epi-phenomenon in both conditions. This is supported by the notion that brain-resident immune cells including microglia and brain-resident CD4+ T cells are essential for neuronal homeostasis and physiological behavior [36][37][38][39]. Moreover, microglia were reported to mediate behavioral deficits in models of depression and anxiety induced by chronic unpredictable stress or early-life inflammation induced by intraperitoneal LPS injection [39][40][41][42]. Involved inflammatory pathways being potential therapeutic targets include microglia–astrocyte crosstalk via glutaminase-1 [4342], the NLRP3 inflammasome [43][44][45] and the clearance of reactive oxygen species (ROS) via silent information regulator 2 homolog 1 (Sirt1)—nuclear factor erythroid 2-related factor 2 (Nrf2)—hemoxygenase 1 (Ho-1)—signaling [45][46][47]. Interestingly, modulation of the BBB is also implicated in the etiology of stress-induced depression and anxiety in chronic social defeat stress. In particular, stress-susceptibility and depression-like behaviors were dependent on downregulation of the tight junction marker claudin-5 (Cldn5) in the hippocampus and nucleus accumbens, which was orchestrated via Tnf and histone deacetylase 1 (Hdac1) and facilitated vascular influx of Il-6 into the brain parenchyma [47][48][49]. As chronic gastrointestinal inflammation was reported to cause BBB tight junction downregulation comparable to social stress, it might also represent a trigger for BBB-mediated depressive-like behavior. Furthermore, non-disruptive BBB alterations may contribute to anxiety-like behavior. Social stress-induced anxiety was mediated by Il1-receptor 1 producing endothelial cells, which were activated by Il-1β-expressing monocytes attracted to the BBB by microglia [5049]. These findings suggest that endothelia closely interact with myeloid cells and may act as a gatekeeper to further develop neuropsychiatric symptoms. A better understanding of non-disruptive BBB changes in IBD is necessary to explore if such mechanisms are present during gastrointestinal inflammation. Though these and other studies strongly imply the relevance of neuroinflammation in depression and anxiety, they do not precisely delineate how neuroinflammation alter the function of neuronal circuits involved in behavioral and emotional regulation to ultimately trigger psychiatric symptoms. In the context of IBD, this might be mediated by several pathways (Figure 2).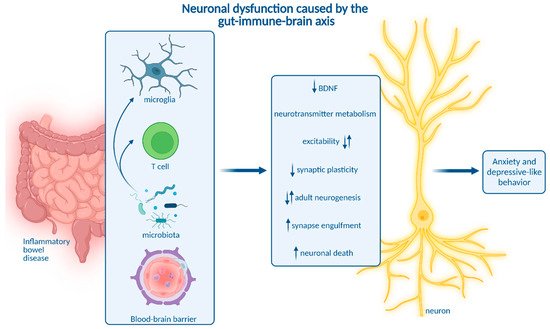
3. Impact of Microbiota on Neuroinflammation and Neuropsychiatric Disease
Having highlighted different routes of transmission from chronic gastrointestinal inflammation into the systemic circulation and into the CNS as well as consecutive neuronal dysfunction and behavioral impairment, reswearchers will shed some light on the role of gut microbiota in the gut-immune-brain interplay. The intestinal microbiota is substantially involved in gastrointestinal inflammation and neuropsychiatric diseases [8380][8481][8582][8677]. Therefore, microbiota and their metabolites are emerging key players in the gut-immune brain axis during IBD. This notion has encouraged several probiotic treatment approaches. Indeed, application of probiotic bacterial strains alleviated DSS-induced colitis, reduced systemic and CNS cytokine levels, induced micro-RNA expression related to restoring inflammation-associated microbiota dysbiosis, and improved depressive and anxiety-like behavior [8783][8884][8985][9086]. However, it is unclear whether effects of probiotic treatments were solely indirect based on the reduction in gut inflammation or also directly interfered with inflammatory gut-to-brain communication or neurons. It is important to note, that changes in the gut microbiota may be cause or consequence of intestinal inflammation and modulate neuropsychiatric symptoms via affecting neuroinflammation, but also exert direct effects on neurons. RWesearchers will therefore highlight different modes of action which affect the CNS during IBD-related dysbiosis. First, microbiota and microbiota-derived molecules actively shape the CNS immune landscape, and the presence of a complex microbiota is essential for microglia activation and function [8985][9187][9288][9389]. Among other CNS-associated myeloid cell types, commensal microbiota strongly influence CP macrophages, while their impact on perivascular and meningeal macrophages is moderate [8985]. Different microbial metabolites were described to modulate microglia. First, microbiota-derived SCFAs signaling via the free fatty acid receptor 2 (Ffar2) were implicated in microglial maturation and function [9288]. A recent differential analysis of distinct SCFAs revealed acetate to be a key regulator of microglial metabolism and phagocytosis [9288]. In the context of multiple sclerosis, the SCFA propionate was shown to induce regulatory Treg activation, whereas pro-inflammatory Th1 and Th17 responses were diminished [9490]. Moreover, bacterial metabolites of dietary tryptophan signal via the aryl hydrocarbon receptor to reduce microglial inflammatory activation of astrocytes [9591]. In the context of IBD, UC patients with comorbid depression or anxiety showed a distinct intestinal bacterial profile linked to reduced blood levels of the metabolites 2ʹ-deoxy-D-ribose and L-pipecolic acid [8380]. Intriguingly, substitution of these metabolites in mice alleviated DSS-induced colitis as well as cytokine levels in the blood and brain, but also ameliorated anxiety and depressive-like behavior [8380]. These findings suggest a role of microbial metabolites in gut-immune-brain communication during IBD. Future studies addressing the modulation of neuroinflammation and behavior in mouse models for experimental colitis by the above-mentioned metabolites will improve people'sur understanding of microbial influence on IBD-related CNS morbidity. In addition to gut bacteria, mucosal fungi were shown to promote Th17 cell activation, which promotes social behavior by direct signaling to neurons via Il-17 [9692]. Collectively, intestinal microbiota are able to modulate systemic and CNS immune responses in IBD and could thereby contribute to IBD-related CNS comorbidity. Besides indirect immune-mediated effects of microbiota and derived metabolites, they have the potential to exert immune-independent effects on neurons via different pathways. Gut bacteria-derived outer membrane vesicles (OMVs) can cross the intestinal barrier and even the BBB, enabling a shuttled transfer of microbial bioactive molecules to the brain [9793]. Interestingly, bacterial OMVs were differentially taken up by neurons with a regionally distinct affinity [9793]. Though their influence on neuronal function is unknown, bacteria-derived OMV cargo uptake may be enhanced in IBD due to gut barrier and BBB dysfunction and may be a complementary part of the gut-to-brain communication.Moreover, intestinal microbiota are involved in neurotransmitter metabolism. Expansion of Bacteroides species contributes to depressive-like behavior and impaired hippocampal neurogenesis by regulating tryptophan and neurotransmitter metabolism [8694]. Interestingly, Bacteroides species were also linked to the development of colitis [9895]. Apart from that, gut microbiota are a major source of the neurotransmitter serotonin. Reduced serotonin abundance as a potential pathogenic mechanism driving depression may be caused by microbial dysbiosis and impaired serotonin production in the gastrointestinal tract [9996].
In addition, gut microbiota influence the expression of micro-RNA (miRNA) in the gut and in different brain regions, which is associated with depression and anxiety in mice [10097][10198]. Interestingly, differential miRNA expression associated with microbiota dysbiosis distinguishes IBD patients and healthy individuals. MiRNAs are thus suggested as biomarkers and promising targets to treat intestinal inflammation [102][160]. Compromised gut microbiota in IBD might therefore contribute to the pathophysiology of concomitant anxiety and depression via affecting neurotransmitter homeostasis and miRNA expression. Besides prototypical neurotransmitters and miRNA, microbial metabolites have the potential to actively signal to neurons. The phenolic compounds phenyl sulfate, pyrocatechol sulfate, and 3-(3-sulfooxyphenyl)propanoic acid as well as indoxyl sulfate were implicated in synaptic remodeling and fear extinction learning [10399]. Moreover, δ-valerobetaine produced by diverse bacterial species modulates inhibitory synaptic transmission and neuronal network activity independent of microglia, preventing age-related decline in cognitive function [104100]. SCFAs are able to act locally in the ENS and enhance enteric neuronal survival and neurogenesis potentially acting via the 5-hydroxytryptamine type 4 (5-HT4) receptor [105101]. SCFAs were also found in the brain, but their levels were not altered during chronic DSS-induced colitis [1817]. In acute DSS-induced colitis, bacteria of the Lachnospiraceae, Ruminococcaceae and Muribaculaceae families were observed to be associated with anxiety and depressive-like behavior. Fecal microbial transfer of colitis-characteristic gut microbiota composition into germ free or antibiotic-treated mice was sufficient to transmit behavioral abnormalities without inducing neuroinflammation, suggesting that immune-independent mechanisms promote microbiota-mediated behavioral alterations in IBD [8481]. Altogether, gut microbiota likely contribute to the development of neuroinflammation and neuropsychiatric symptoms in IBD. Future studies will need to identify essential bacterial strains and the mechanisms they employ to alter neuroinflammation or directly modify neuronal function specifically during chronic gastrointestinal inflammation.
References
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. Sigrid Breit; Aleksandra Kupferberg; Gerhard Rogler; Gregor Hasler; Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Frontiers in Psychiatry 2018, 9, 44, 10.3389/fpsyt.2018.00044.
- Ichiki, T.; Wang, T.; Kennedy, A.; Pool, A.-H.; Ebisu, H.; Anderson, D.J.; Oka, Y. Sensory representation and detection mechanisms of gut osmolality change. Nature 2022, 602, 468–474. Takako Ichiki; Tongtong Wang; Ann Kennedy; Allan-Hermann Pool; Haruka Ebisu; David J. Anderson; Yuki Oka; Sensory representation and detection mechanisms of gut osmolality change. Nature 2022, 602, 468-474, 10.1038/s41586-021-04359-5.
- Goswami, C.; Iwasaki, Y.; Yada, T. Short-chain fatty acids suppress food intake by activating vagal afferent neurons. J. Nutr. Biochem. 2018, 57, 130–135. Chayon Goswami; Yusaku Iwasaki; Toshihiko Yada; Short-chain fatty acids suppress food intake by activating vagal afferent neurons. The Journal of Nutritional Biochemistry 2018, 57, 130-135, 10.1016/j.jnutbio.2018.03.009.
- Browning, K.N.; Verheijden, S.; Boeckxstaens, G.E. The Vagus Nerve in Appetite Regulation, Mood, and Intestinal Inflammation. Gastroenterology 2017, 152, 730–744. Kirsteen N. Browning; Simon Verheijden; Guy E. Boeckxstaens; The Vagus Nerve in Appetite Regulation, Mood, and Intestinal Inflammation. Gastroenterology 2017, 152, 730-744, 10.1053/j.gastro.2016.10.046.
- Steinberg, B.E.; Silverman, H.A.; Robbiati, S.; Gunasekaran, M.K.; Tsaava, T.; Battinelli, E.; Stiegler, A.; Bouton, C.E.; Chavan, S.S.; Tracey, K.J.; et al. Cytokine-specific Neurograms in the Sensory Vagus Nerve. Bioelectron. Med. 2016, 3, 7–17. Benjamin E. Steinberg; Harold A. Silverman; Sergio Robbiati; Manoj K. Gunasekaran; Téa Tsaava; Emily Battinelli; Andrew Stiegler; Chad E. Bouton; Sangeeta S. Chavan; Kevin J. Tracey; et al.Patricio T. Huerta Cytokine-specific Neurograms in the Sensory Vagus Nerve. Bioelectronic Medicine 2016, 3, 7-17, 10.15424/bioelectronmed.2016.00007.
- Watkins, L.R.; Goehler, L.E.; Relton, J.K.; Tartaglia, N.; Silbert, L.; Martin, D.; Maier, S.F. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: Evidence for vagal mediation of immune-brain communication. Neurosci. Lett. 1995, 183, 27–31. Linda R. Watkins; Lisa E. Goehler; Jane K. Relton; Nicole Tartaglia; Lee Silbert; David Martin; Steven F. Maier; Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neuroscience Letters 1995, 183, 27-31, 10.1016/0304-3940(94)11105-r.
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic Gastrointestinal Inflammation Induces Anxiety-Like Behavior and Alters Central Nervous System Biochemistry in Mice. Gastroenterology 2010, 139, 2102–2112.e1. Premysl Bercik; Elena F. Verdu; Jane A. Foster; Joseph Macri; Murray Potter; Xiaxing Huang; Paul Malinowski; Wendy Jackson; Patricia Blennerhassett; Karen A. Neufeld; et al.Jun LuWaliul I. KhanIrene Corthesy–TheulazChristine CherbutGabriela E. BergonzelliStephen M. Collins Chronic Gastrointestinal Inflammation Induces Anxiety-Like Behavior and Alters Central Nervous System Biochemistry in Mice. Gastroenterology 2010, 139, 2102-2112.e1, 10.1053/j.gastro.2010.06.063.
- Sun, P.; Zhou, K.; Wang, S.; Li, P.; Chen, S.; Lin, G.; Zhao, Y.; Wang, T. Involvement of MAPK/NF-κB signaling in the activation of the cholinergic anti-inflammatory pathway in experimental colitis by chronic vagus nerve stimulation. PLoS ONE 2013, 8, e69424. Peng Sun; Kewen Zhou; Sheng Wang; Ping Li; Sijuan Chen; Guiping Lin; Yan Zhao; Tinghuai Wang; Involvement of MAPK/NF-κB Signaling in the Activation of the Cholinergic Anti-Inflammatory Pathway in Experimental Colitis by Chronic Vagus Nerve Stimulation. PLOS ONE 2013, 8, e69424, 10.1371/journal.pone.0069424.
- Sinniger, V.; Pellissier, S.; Fauvelle, F.; Trocmé, C.; Hoffmann, D.; Vercueil, L.; Cracowski, J.L.; David, O.; Bonaz, B. A 12-month pilot study outcomes of vagus nerve stimulation in Crohn’s disease. Neurogastroenterol. Motil. 2020, 32, e13911. Charles Ibeakanma; Stephen Vanner; TNFα is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut 2010, 59, 612-621, 10.1136/gut.2009.190439.
- Ibeakanma, C.; Vanner, S.J. TNI± is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut 2010, 59, 612–621. Andreas Hess; Julie Roesch; Marc Saake; Marina Sergeeva; Simon Hirschmann; Helmut Neumann; Arnd Dörfler; Markus Friedrich Neurath; Raja Atreya; Functional Brain Imaging Reveals Rapid Blockade of Abdominal Pain Response Upon Anti-TNF Therapy in Crohn’s Disease. Gastroenterology 2015, 149, 864-866, 10.1053/j.gastro.2015.05.063.
- Hess, A.; Roesch, J.; Saake, M.; Sergeeva, M.; Hirschmann, S.; Neumann, H.; Dörfler, A.; Neurath, M.F.; Atreya, R. Functional Brain Imaging Reveals Rapid Blockade of Abdominal Pain Response Upon Anti-TNF Therapy in Crohn’s Disease. Gastroenterology 2015, 149, 864–866. Britta Engelhardt; Peter Vajkoczy; Roy O Weller; The movers and shapers in immune privilege of the CNS. Nature Immunology 2017, 18, 123-131, 10.1038/ni.3666.
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. Ian Galea; The blood–brain barrier in systemic infection and inflammation. Cellular & Molecular Immunology 2021, 18, 2489-2501, 10.1038/s41423-021-00757-x.
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. Ilaria Spadoni; Elena Zagato; Alice Bertocchi; Roberta Paolinelli; Edina Hot; Antonio Di Sabatino; Flavio Caprioli; Luca Bottiglieri; Amanda Oldani; Giuseppe Viale; et al.Giuseppe PennaElisabetta DejanaMaria Rescigno A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830-834, 10.1126/science.aad0135.
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. Aravinthan Varatharaj; Ian Galea; The blood-brain barrier in systemic inflammation. Brain, Behavior, and Immunity 2017, 60, 1-12, 10.1016/j.bbi.2016.03.010.
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. Ying Han; Tong Zhao; Xiang Cheng; Ming Zhao; Sheng-Hui Gong; Yong-Qi Zhao; Hai-Tao Wu; Ming Fan; Ling-Ling Zhu; Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neuroscience Bulletin 2018, 34, 1058-1066, 10.1007/s12264-018-0288-5.
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.-H.; Zhao, Y.-Q.; Wu, H.-T.; Fan, M.; Zhu, L.-L. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci. Bull. 2018, 34, 1058–1066. Ying Han; Liping Ding; Xiang Cheng; Ming Zhao; Tong Zhao; Liang Guo; Xinyang Li; Yanan Geng; Ming Fan; Hong Liao; et al.Lingling Zhu Hypoxia Augments Cerebral Inflammation in a Dextran Sulfate Sodium-Induced Colitis Mouse Model. Frontiers in Cellular Neuroscience 2020, 14, 611764, 10.3389/fncel.2020.611764.
- Han, Y.; Ding, L.; Cheng, X.; Zhao, M.; Zhao, T.; Guo, L.; Li, X.; Geng, Y.; Fan, M.; Liao, H.; et al. Hypoxia Augments Cerebral Inflammation in a Dextran Sulfate Sodium-Induced Colitis Mouse Model. Front. Cell. Neurosci. 2020, 14, 611764. Jonathon Mitchell; Su Jin Kim; Cody Howe; Seulah Lee; Ji Yun Her; Marisa Patel; Gayoung Kim; Jaewon Lee; Eunok Im; Sang Hoon Rhee; et al. Chronic Intestinal Inflammation Suppresses Brain Activity by Inducing Neuroinflammation in Mice. The American Journal of Pathology 2021, 192, 72-86, 10.1016/j.ajpath.2021.09.006.
- Mitchell, J.; Kim, S.J.; Howe, C.; Lee, S.; Her, J.Y.; Patel, M.; Kim, G.; Lee, J.; Im, E.; Rhee, S.H. Chronic Intestinal Inflammation Suppresses Brain Activity by Inducing Neuroinflammation in Mice. Am. J. Pathol. 2022, 192, 72–86. Sara Carloni; Alice Bertocchi; Sara Mancinelli; Martina Bellini; Marco Erreni; Antonella Borreca; Daniele Braga; Silvia Giugliano; Alessandro M. Mozzarelli; Daria Manganaro; et al.Daniel Fernandez PerezFederico ColomboAntonio Di SabatinoDiego PasiniGiuseppe PennaMichela MatteoliSimona LodatoMaria Rescigno Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439-448, 10.1126/science.abc6108.
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. Xiaolin Zhao; Peng Liang; Jin Liu; Haixia Jiang; Xiaoshuai Fan; Guo Chen; Cheng Zhou; Elevation of arachidonoylethanolamide levels by activation of the endocannabinoid system protects against colitis and ameliorates remote organ lesions in mice. Experimental and Therapeutic Medicine 2017, 14, 5664-5670, 10.3892/etm.2017.5222.
- Zhao, X.; Liang, P.; Liu, J.; Jiang, H.; Fan, X.; Chen, G.; Zhou, C. Elevation of arachidonoylethanolamide levels by activation of the endocannabinoid system protects against colitis and ameliorates remote organ lesions in mice. Exp. Ther. Med. 2017, 14, 5664–5670. Christopher A. Hathaway; Caroline B. Appleyard; William H. Percy; John L. Williams; Experimental colitis increases blood-brain barrier permeability in rabbits. American Journal of Physiology-Gastrointestinal and Liver Physiology 1999, 276, G1174-G1180, 10.1152/ajpgi.1999.276.5.g1174.
- Hathaway, C.A.; Appleyard, C.B.; Percy, W.H.; Williams, J.L. Experimental colitis increases blood-brain barrier permeability in rabbits. Am. J. Physiol.-Gastrointest. Liver Physiol. 1999, 276, G1174–G1180. S. S. Natah; A. Mouihate; Q. J. Pittman; K. A. Sharkey; Disruption of the blood-brain barrier during TNBS colitis. Neurogastroenterology & Motility 2005, 17, 433-446, 10.1111/j.1365-2982.2005.00654.x.
- Natah, S.S.; Mouihate, A.; Pittman, Q.J.; Sharkey, K.A. Disruption of the blood-brain barrier during TNBS colitis. Neurogastroenterol. Motil. 2005, 17, 433–446. Sarah E. Barnes; Kristy A. Zera; Geoffrey T. Ivison; Marion S. Buckwalter; Edgar G. Engleman; Brain profiling in murine colitis and human epilepsy reveals neutrophils and TNFα as mediators of neuronal hyperexcitability. Journal of Neuroinflammation 2021, 18, 1-13, 10.1186/s12974-021-02262-4.
- Barnes, S.E.; Zera, K.A.; Ivison, G.T.; Buckwalter, M.S.; Engleman, E.G. Brain profiling in murine colitis and human epilepsy reveals neutrophils and TNFα as mediators of neuronal hyperexcitability. J. Neuroinflamm. 2021, 18, 199. Sarah Talley; Rasa Valiauga; Lillian Anderson; Abigail R. Cannon; Mashkoor A. Choudhry; Edward M. Campbell; DSS-induced inflammation in the colon drives a proinflammatory signature in the brain that is ameliorated by prophylactic treatment with the S100A9 inhibitor paquinimod. Journal of Neuroinflammation 2021, 18, 1-14, 10.1186/s12974-021-02317-6.
- Talley, S.; Valiauga, R.; Anderson, L.; Cannon, A.R.; Choudhry, M.A.; Campbell, E.M. DSS-induced inflammation in the colon drives a proinflammatory signature in the brain that is ameliorated by prophylactic treatment with the S100A9 inhibitor paquinimod. J. Neuroinflamm. 2021, 18, 263. Thomas Blank; Claudia N. Detje; Alena Spieß; Nora Hagemeyer; Stefanie M. Brendecke; Jakob Wolfart; Ori Staszewski; Tanja Zöller; Ismini Papageorgiou; Justus Schneider; et al.Ricardo Paricio-MontesinosUlrich L.M. EiselDenise Manahan-VaughanStephan JansenStefan LienenklausBao LuYumiko ImaiMarcus MüllerSusan E. GoelzDarren P. BakerMarkus SchwaningerOliver KannMathias HeikenwalderUlrich KalinkeMarco Prinz Brain Endothelial- and Epithelial-Specific Interferon Receptor Chain 1 Drives Virus-Induced Sickness Behavior and Cognitive Impairment. Immunity 2016, 44, 901-912, 10.1016/j.immuni.2016.04.005.
- Blank, T.; Detje, C.N.; Spieß, A.; Hagemeyer, N.; Brendecke, S.M.; Wolfart, J.; Staszewski, O.; Zöller, T.; Papageorgiou, I.; Schneider, J.; et al. Brain Endothelial- and Epithelial-Specific Interferon Receptor Chain 1 Drives Virus-Induced Sickness Behavior and Cognitive Impairment. Immunity 2016, 44, 901–912. Hanadie Yousef; Cathrin J. Czupalla; Davis Lee; Michelle B. Chen; Ashley Burke; Kristy Zera; Judith Zandstra; Elisabeth Berber; Benoit Lehallier; Vidhu Mathur; et al.Ramesh V. NairLiana BonannoAndrew C. YangTodd PetersonHusein HadeibaTaylor MerkelJakob KörbelinMarkus SchwaningerMarion S. BuckwalterStephen R. QuakeEugene C. ButcherTony Wyss-Coray Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nature Medicine 2019, 25, 988-1000, 10.1038/s41591-019-0440-4.
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000. Noha Althubaity; Julia Schubert; Daniel Martins; Tayyabah Yousaf; Maria A. Nettis; Valeria Mondelli; Carmine Pariante; Neil A. Harrison; Edward T. Bullmore; Danai Dima; et al.Federico E. TurkheimerMattia Veronese Choroid plexus enlargement is associated with neuroinflammation and reduction of blood brain barrier permeability in depression. NeuroImage: Clinical 2021, 33, 102926, 10.1016/j.nicl.2021.102926.
- Althubaity, N.; Schubert, J.; Martins, D.; Yousaf, T.; Nettis, M.A.; Mondelli, V.; Pariante, C.; Harrison, N.A.; Bullmore, E.T.; Dima, D.; et al. Choroid plexus enlargement is associated with neuroinflammation and reduction of blood brain barrier permeability in depression. NeuroImage Clin. 2022, 33, 102926. Sriram Balusu; Elien Van Wonterghem; Riet De Rycke; Koen Raemdonck; Stephan Stremersch; Kris Gevaert; Marjana Brkic; Delphine Demeestere; Valerie Vanhooren; An Hendrix; et al.Claude LibertRoosmarijn E Vandenbroucke Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus‐derived extracellular vesicles. EMBO Molecular Medicine 2016, 8, 1162-1183, 10.15252/emmm.201606271.
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus—derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. Hannah Van Hove; Liesbet Martens; Isabelle Scheyltjens; Karen De Vlaminck; Ana Rita Pombo Antunes; Sofie De Prijck; Niels Vandamme; Sebastiaan De Schepper; Gert Van Isterdael; Charlotte L. Scott; et al.Jeroen AertsGeert BerxGuy E. BoeckxstaensRoosmarijn E. VandenbrouckeLars VereeckeDiederik MoecharsMartin GuilliamsJo Van GinderachterYvan SaeysKiavash Movahedi A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nature Neuroscience 2019, 22, 1021-1035, 10.1038/s41593-019-0393-4.
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Antunes, A.R.P.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. Kuti Baruch; Aleksandra Deczkowska; Eyal David; Joseph M. Castellano; Omer Miller; Alexander Kertser; Tamara Berkutzki; Zohar Barnett-Itzhaki; Dana Bezalel; Tony Wyss-Coray; et al.Ido AmitMichal Schwartz Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89-93, 10.1126/science.1252945.
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. Elaine Dempsey; Áine Abautret-Daly; Neil G. Docherty; Carlos Medina; Andrew Harkin; Persistent central inflammation and region specific cellular activation accompany depression- and anxiety-like behaviours during the resolution phase of experimental colitis. Brain, Behavior, and Immunity 2019, 80, 616-632, 10.1016/j.bbi.2019.05.007.
- Dempsey, E.; Abautret-Daly, Á.; Docherty, N.G.; Medina, C.; Harkin, A. Persistent central inflammation and region specific cellular activation accompany depression- and anxiety-like behaviours during the resolution phase of experimental colitis. Brain Behav. Immun. 2019, 80, 616–632. Anthony J. Filiano; Yang Xu; Nicholas Tustison; Rachel L. Marsh; Wendy Baker; Igor Smirnov; Christopher C. Overall; Sachin P. Gadani; Stephen Turner; Zhiping Weng; et al.Sayeda Najamussahar PeerzadeHao ChenKevin S. LeeMichael M. ScottMark P. BeenhakkerVladimir LitvakJonathan Kipnis Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 2016, 535, 425-429, 10.1038/nature18626.
- Filiano, A.J.; Xu, Y.; Tustison, N.; Marsh, R.L.; Baker, W.; Smirnov, I.; Overall, C.C.; Gadani, S.P.; Turner, S.; Weng, Z.; et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 2016, 535, 425–429. Kalil Alves De Lima; Justin Rustenhoven; Sandro Da Mesquita; Morgan Wall; Andrea Francesca Salvador; Igor Smirnov; Guilherme Martelossi Cebinelli; Tornike Mamuladze; Wendy Baker; Zach Papadopoulos; et al.Maria Beatriz LopesWilliam Sam CaoXinmin Simon XieJasmin HerzJonathan Kipnis Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nature Immunology 2020, 21, 1421-1429, 10.1038/s41590-020-0776-4.
- De Lima, K.A.; Rustenhoven, J.; Da Mesquita, S.; Wall, M.; Salvador, A.F.; Smirnov, I.; Cebinelli, G.M.; Mamuladze, T.; Baker, W.; Papadopoulos, Z.; et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat. Immunol. 2020, 21, 1421–1429. David Brea; Carrie Poon; Corinne Benakis; Gabrielle Lubitz; Michelle Murphy; Costantino Iadecola; Josef Anrather; Stroke affects intestinal immune cell trafficking to the central nervous system. Brain, Behavior, and Immunity 2021, 96, 295-302, 10.1016/j.bbi.2021.05.008.
- Brea, D.; Poon, C.; Benakis, C.; Lubitz, G.; Murphy, M.; Iadecola, C.; Anrather, J. Stroke affects intestinal immune cell trafficking to the central nervous system. Brain Behav. Immun. 2021, 96, 295–302. Pia Kivisäkk; Barbara Tucky; Tao Wei; James J Campbell; Richard M Ransohoff; Human cerebrospinal fluid contains CD4+ memory T cells expressing gut- or skin-specific trafficking determinants: relevance for immunotherapy. BMC Immunology 2006, 7, 14-14, 10.1186/1471-2172-7-14.
- Kivisäkk, P.; Tucky, B.; Wei, T.; Campbell, J.J.; Ransohoff, R.M. Human cerebrospinal fluid contains CD4+ memory T cells expressing gut- or skin-specific trafficking determinants: Relevance for immunotherapy. BMC Immunol. 2006, 7, 14. Xiao-Fei He; Li-Li Li; Wen-Biao Xian; Ming-Yue Li; Li-Ying Zhang; Jing-Hui Xu; Zhong Pei; Hai-Qing Zheng; Xi-Quan Hu; Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. Journal of Neuroinflammation 2021, 18, 153, 10.1186/s12974-021-02199-8.
- He, X.-F.; Li, L.-L.; Xian, W.-B.; Li, M.-Y.; Zhang, L.-Y.; Xu, J.-H.; Pei, Z.; Zheng, H.-Q.; Hu, X.-Q. Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. J. Neuroinflamm. 2021, 18, 153. Ana Badimon; Hayley J. Strasburger; Pinar Ayata; Xinhong Chen; Aditya Nair; Ako Ikegami; Philip Hwang; Andrew T. Chan; Steven M. Graves; Joseph O. Uweru; et al.Carola LedderoseMunir Gunes KutluMichael A. WheelerAnat KahanMasago IshikawaYing-Chih WangYong-Hwee E. LohJean X. JiangD. James SurmeierSimon C. RobsonWolfgang G. JungerRobert SebraErin S. CalipariPaul J. KennyUkpong B. EyoMarco ColonnaFrancisco J. QuintanaHiroaki WakeViviana GradinaruAnne Schaefer Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417-423, 10.1038/s41586-020-2777-8.
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. Christopher N. Parkhurst; Guang Yang; Ipe Ninan; Jeffrey N. Savas; John R. Yates; Juan J. Lafaille; Barbara L. Hempstead; Dan R. Littman; Wen-Biao Gan; Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596-1609, 10.1016/j.cell.2013.11.030.
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. Emanuela Pasciuto; Oliver T. Burton; Carlos P. Roca; Vasiliki Lagou; Wenson D. Rajan; Tom Theys; Renzo Mancuso; Raul Y. Tito; Lubna Kouser; Zsuzsanna Callaerts-Vegh; et al.Alerie Guzman de la FuenteTeresa PrezzemoloLoriana G. MascaliAleksandra BrajicCarly E. WhyteLidia YshiiAnna Martinez-MurianaMichelle NaughtonAndrew YoungAlena MoudraPierre LemaitreSuresh PoovathingalJeroen RaesBart De StrooperDenise C. FitzgeraldJames DooleyAdrian Liston Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625-640.e24, 10.1016/j.cell.2020.06.026.
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.e24. Anna M. Klawonn; Michael Fritz; Silvia Castany; Marco Pignatelli; Carla Canal; Fredrik Similä; Hugo A. Tejeda; Julia Levinsson; Maarit Jaarola; Johan Jakobsson; et al.Juan HidalgoMarkus HeiligAntonello BonciDavid Engblom Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021, 54, 225-234.e6, 10.1016/j.immuni.2020.12.016.
- Klawonn, A.M.; Fritz, M.; Castany, S.; Pignatelli, M.; Canal, C.; Similä, F.; Tejeda, H.A.; Levinsson, J.; Jaarola, M.; Jakobsson, J.; et al. Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 2021, 54, 225–234.e6. Peng Cao; Changmao Chen; An Liu; Qinghong Shan; Xia Zhu; Chunhui Jia; Xiaoqi Peng; Mingjun Zhang; Zahra Farzinpour; Wenjie Zhou; et al.Haitao WangJiang-Ning ZhouXiaoyuan SongLiecheng WangWenjuan TaoChangjian ZhengYan ZhangYu-Qiang DingYan JinLin XuZhi Zhang Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573-2589.e9, 10.1016/j.neuron.2021.06.012.
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e9. Eric S. Wohleb; Rosemarie Terwilliger; Catharine H. Duman; Ronald S. Duman; Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biological Psychiatry 2017, 83, 38-49, 10.1016/j.biopsych.2017.05.026.
- Wohleb, E.S.; Terwilliger, R.; Duman, C.H.; Duman, R.S. Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol. Psychiatry 2018, 83, 38–49. Chenhui Ji; Yalin Tang; Yanyan Zhang; Congcong Li; Huazheng Liang; Lu Ding; Xiaohuan Xia; Lize Xiong; Xin-Rui Qi; Jialin C. Zheng; et al. Microglial glutaminase 1 deficiency mitigates neuroinflammation associated depression. Brain, Behavior, and Immunity 2021, 99, 231-245, 10.1016/j.bbi.2021.10.009.
- Ji, C.; Tang, Y.; Zhang, Y.; Li, C.; Liang, H.; Ding, L.; Xia, X.; Xiong, L.; Qi, X.-R.; Zheng, J.C. Microglial glutaminase 1 deficiency mitigates neuroinflammation associated depression. Brain Behav. Immun. 2022, 99, 231–245. Weifen Li; Tahir Ali; Kaiwu He; Zizhen Liu; Fawad Ali Shah; Qingguo Ren; Yan Liu; Anlong Jiang; Shupeng Li; Ibrutinib alleviates LPS-induced neuroinflammation and synaptic defects in a mouse model of depression. Brain, Behavior, and Immunity 2020, 92, 10-24, 10.1016/j.bbi.2020.11.008.
- Li, W.; Ali, T.; He, K.; Liu, Z.; Shah, F.A.; Ren, Q.; Liu, Y.; Jiang, A.; Li, S. Ibrutinib alleviates LPS-induced neuroinflammation and synaptic defects in a mouse model of depression. Brain Behav. Immun. 2021, 92, 10–24. Fernanda N. Kaufmann; Ana Paula Costa; Gabriele Ghisleni; Alexandre Diaz; Ana Lúcia Rodrigues; Hugo Peluffo; Manuella P. Kaster; NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain, Behavior, and Immunity 2017, 64, 367-383, 10.1016/j.bbi.2017.03.002.
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.; Rodrigues, A.L.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain Behav. Immun. 2017, 64, 367–383. Ruozhi Dang; Mingyang Wang; Xinhui Li; Haiyang Wang; Lanxiang Liu; Qingyuan Wu; Jianting Zhao; Ping Ji; Lianmei Zhong; Julio Licinio; et al.Peng Xie Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. Journal of Neuroinflammation 2022, 19, 1-29, 10.1186/s12974-022-02400-6.
- Dang, R.; Wang, M.; Li, X.; Wang, H.; Liu, L.; Wu, Q.; Zhao, J.; Ji, P.; Zhong, L.; Licinio, J.; et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J. Neuroinflamm. 2022, 19, 41. Tahir Ali; Qiang Hao; Najeeb Ullah; Shafiq Ur Rahman; Fawad Ali Shah; Kaiwu He; Chengyou Zheng; Weifen Li; Iram Murtaza; Yang Li; et al.Yuhua JiangZhen TanShupeng Li Melatonin Act as an Antidepressant via Attenuation of Neuroinflammation by Targeting Sirt1/Nrf2/HO-1 Signaling. Frontiers in Molecular Neuroscience 2020, 13, 96, 10.3389/fnmol.2020.00096.
- Ali, T.; Hao, Q.; Ullah, N.; Rahman, S.U.; Shah, F.A.; He, K.; Zheng, C.; Li, W.; Murtaza, I.; Li, Y.; et al. Melatonin Act as an Antidepressant via Attenuation of Neuroinflammation by Targeting Sirt1/Nrf2/HO-1 Signaling. Front. Mol. Neurosci. 2020, 13, 96. Katarzyna A. Dudek; Laurence Dion-Albert; Manon Lebel; Katherine LeClair; Simon Labrecque; Ellen Tuck; Carmen Ferrer Perez; Sam A. Golden; Carol Tamminga; Gustavo Turecki; et al.Naguib MechawarScott J. RussoCaroline Menard Molecular adaptations of the blood–brain barrier promote stress resilience vs. depression. Proceedings of the National Academy of Sciences 2020, 117, 3326-3336, 10.1073/pnas.1914655117.
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Perez, C.F.; Golden, S.A.; Tamminga, C.; Turecki, G.; et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. USA 2020, 117, 3326–3336. Caroline Menard; Madeline L. Pfau; Georgia Hodes; Veronika Kana; Victoria X. Wang; Sylvain Bouchard; Aki Takahashi; Meghan E. Flanigan; Hossein Aleyasin; Katherine B. LeClair; et al.William G. JanssenBenoit LabontéEric M. PariseZachary S. LorschSam A. GoldenMitra HeshmatiCarol TammingaGustavo TureckiMatthew CampbellZahi A. FayadCheuk Ying TangMiriam MeradScott J. Russo Social stress induces neurovascular pathology promoting depression. Nature Neuroscience 2017, 20, 1752-1760, 10.1038/s41593-017-0010-3.
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. D B McKim; M D Weber; A Niraula; C M Sawicki; X Liu; B L Jarrett; K Ramirez-Chan; Y Wang; R M Roeth; A D Sucaldito; et al.C G SobolN QuanJ F SheridanJ P Godbout Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Molecular Psychiatry 2017, 23, 1421-1431, 10.1038/mp.2017.64.
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 2018, 23, 1421–1431. Zhi-Heng Zheng; Jiang-Long Tu; Xiao-Han Li; Qing Hua; Wei-Zhu Liu; Yu Liu; Bing-Xing Pan; Ping Hu; Wen-Hua Zhang; Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain, Behavior, and Immunity 2020, 91, 505-518, 10.1016/j.bbi.2020.11.007.
- Zheng, Z.-H.; Tu, J.-L.; Li, X.-H.; Hua, Q.; Liu, W.-Z.; Liu, Y.; Pan, B.-X.; Hu, P.; Zhang, W.-H. Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain Behav. Immun. 2021, 91, 505–518. Kiarash Riazi; Michael A. Galic; J. Brent Kuzmiski; Winnie Ho; Keith A. Sharkey; Quentin J. Pittman; Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proceedings of the National Academy of Sciences 2008, 105, 17151-17156, 10.1073/pnas.0806682105.
- Riazi, K.; Galic, M.A.; Kuzmiski, J.B.; Ho, W.; Sharkey, K.A.; Pittman, Q.J. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 17151–17156. Colin F. Craig; Rhiannon T. Filippone; Rhian Stavely; Joel C. Bornstein; Vasso Apostolopoulos; Kulmira Nurgali; Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. Journal of Neuroinflammation 2022, 19, 1-30, 10.1186/s12974-021-02354-1.
- Craig, C.F.; Filippone, R.T.; Stavely, R.; Bornstein, J.C.; Apostolopoulos, V.; Nurgali, K. Neuroinflammation as an etiological trigger for depression comorbid with inflammatory bowel disease. J. Neuroinflamm. 2022, 19, 4. Jean-Eric Ghia; Nan Li; Huaqing Wang; Matthew Collins; Yikang Deng; Rami El-Sharkawy; Francine Côté; Jacques Mallet; Waliul I. Khan; Serotonin Has a Key Role in Pathogenesis of Experimental Colitis. Gastroenterology 2009, 137, 1649-1660, 10.1053/j.gastro.2009.08.041.
- Ghia, J.E.; Li, N.; Wang, H.; Collins, M.; Deng, Y.; El-Sharkawy, R.T.; Côté, F.; Mallet, J.; Khan, W.I. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology 2009, 137, 1649–1660. Sharif Shajib; Adriana Baranov; Waliul Islam Khan; Diverse Effects of Gut-Derived Serotonin in Intestinal Inflammation. ACS Chemical Neuroscience 2017, 8, 920-931, 10.1021/acschemneuro.6b00414.
- Shajib, M.S.; Baranov, A.; Khan, W.I. Diverse Effects of Gut-Derived Serotonin in Intestinal Inflammation. ACS Chem. Neurosci. 2017, 8, 920–931. Marie Skov Kristensen; Thora Majlund Kjærulff; Annette Kjær Ersbøll; Anders Green; Jesper Hallas; Lau Thygesen; The Influence of Antidepressants on the Disease Course Among Patients With Crohn’s Disease and Ulcerative Colitis—A Danish Nationwide Register–Based Cohort Study. Inflammatory Bowel Diseases 2018, 25, 886-893, 10.1093/ibd/izy367.
- Kristensen, M.S.; Kjærulff, T.M.; Ersbøll, A.K.; Green, A.; Hallas, J.; Thygesen, L.C. The Influence of Antidepressants on the Disease Course among Patients with Crohn’s Disease and Ulcerative Colitis-A Danish Nationwide Register-Based Cohort Study. Inflamm. Bowel Dis. 2019, 25, 886–893. Li-Ming Chen; Chun-Hui Bao; Yu Wu; Shi-Hua Liang; Di Di Wang; Lu-Yi Wu; Yan Huang; Hui-Rong Liu; Huan-Gan Wu; Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. Journal of Neuroinflammation 2021, 18, 1-13, 10.1186/s12974-021-02175-2.
- Chen, L.-M.; Bao, C.-H.; Wu, Y.; Liang, S.-H.; Wang, D.; Wu, L.-Y.; Huang, Y.; Liu, H.-R.; Wu, H.-G. Tryptophan-kynurenine metabolism: A link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflamm. 2021, 18, 135. Hoda M. Sroor; Ahmed M. Hassan; Geraldine Zenz; Paulina Valadez-Cosmes; Aitak Farzi; Peter Holzer; Amany El-Sharif; Fatma Al-Zahraa M. Gomaa; Julia Kargl; Florian Reichmann; et al. Experimental colitis reduces microglial cell activation in the mouse brain without affecting microglial cell numbers. Scientific Reports 2019, 9, 1-12, 10.1038/s41598-019-56859-0.
- Sroor, H.M.; Hassan, A.M.; Zenz, G.; Valadez-Cosmes, P.; Farzi, A.; Holzer, P.; El-Sharif, A.; Gomaa, F.A.-Z.M.; Kargl, J.; Reichmann, F. Experimental colitis reduces microglial cell activation in the mouse brain without affecting microglial cell numbers. Sci. Rep. 2019, 9, 20217. Patrick Süß; Microglia in Alzheimer’s Disease. Current Alzheimer Research 2020, 17, 29-43, 10.2174/1567205017666200212155234.
- Süß, P.; Schlachetzki, J.C.M. Microglia in Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 29–43. Amanda Crider; Tami Feng; Chirayu D. Pandya; Talisha Davis; Ashwati Nair; Anthony O. Ahmed; Babak Baban; Gustavo Turecki; Anilkumar Pillai; Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain, Behavior, and Immunity 2018, 70, 246-256, 10.1016/j.bbi.2018.03.004.
- Crider, A.; Feng, T.; Pandya, C.D.; Davis, T.; Nair, A.; Ahmed, A.O.; Baban, B.; Turecki, G.; Pillai, A. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav. Immun. 2018, 70, 246–256. Jessica L. Bolton; Annabel K. Short; Shivashankar Othy; Cassandra L. Kooiker; Manlin Shao; Benjamin G. Gunn; Jaclyn Beck; Xinglong Bai; Stephanie M. Law; Julie C. Savage; et al.Jeremy J. LambertDelia BelelliMarie-Ève TremblayMichael D. CahalanTallie Z. Baram Early stress-induced impaired microglial pruning of excitatory synapses on immature CRH-expressing neurons provokes aberrant adult stress responses. Cell Reports 2022, 38, 110600, 10.1016/j.celrep.2022.110600.
- Bolton, J.L.; Short, A.K.; Othy, S.; Kooiker, C.L.; Shao, M.; Gunn, B.G.; Beck, J.; Bai, X.; Law, S.M.; Savage, J.C.; et al. Early stress-induced impaired microglial pruning of excitatory synapses on immature CRH-expressing neurons provokes aberrant adult stress responses. Cell Rep. 2022, 38, 110600. B L Jacobs; H van Praag; F H Gage; Adult brain neurogenesis and psychiatry: a novel theory of depression. Molecular Psychiatry 2000, 5, 262-269, 10.1038/sj.mp.4000712.
- Vadodaria, K.C.; Gage, F.H. SnapShot: Adult Hippocampal Neurogenesis. Cell 2014, 156, 1114–1114.e1. Tomohisa Toda; Sarah L. Parylak; Sara B. Linker; Fred H. Gage; The role of adult hippocampal neurogenesis in brain health and disease. Molecular Psychiatry 2018, 24, 67-87, 10.1038/s41380-018-0036-2.
- Jacobs, B.L.; van Praag, H.; Gage, F.H. Adult brain neurogenesis and psychiatry: A novel theory of depression. Mol. Psychiatry 2000, 5, 262–269. Amanda Sierra; Juan Manuel Encinas; Juan José Peña Deudero; Jessica Chancey; Grigori Enikolopov; Linda Wadiche; Stella E. Tsirka; Mirjana Maletic-Savatic; Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483-495, 10.1016/j.stem.2010.08.014.
- Toda, T.; Parylak, S.L.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. Robert E. Iosif; Christine T. Ekdahl; Henrik Ahlenius; Cornelis J. H. Pronk; Sara Bonde; Zaal Kokaia; Sten Eirik Waelgaard Jacobsen; Olle Lindvall; Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. The Journal of Neuroscience 2006, 26, 9703-9712, 10.1523/jneurosci.2723-06.2006.
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.; Enikolopov, G.; Wadiche, L.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. Inbal Goshen; T Kreisel; O Ben-Menachem-Zidon; Tamar Licht; J Weidenfeld; T Ben-Hur; R Yirmiya; Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Molecular Psychiatry 2007, 13, 717-728, 10.1038/sj.mp.4002055.
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.W.; Lindvall, O. Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. J. Neurosci. 2006, 26, 9703–9712. Michelle L. Monje; Hiroki Toda; Theo D. Palmer; Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760-1765, 10.1126/science.1088417.
- Goshen, I.; Kreisel, T.; Ben-Menachem-Zidon, O.; Licht, T.; Weidenfeld, J.; Ben-Hur, T.; Yirmiya, R. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 2007, 13, 717–728. Maiko Tatsuki; Reiko Hatori; Tomoko Nakazawa; Takashi Ishige; Tomoko Hara; Seiichi Kagimoto; Takeshi Tomomasa; Hirokazu Arakawa; Takumi Takizawa; Serological cytokine signature in paediatric patients with inflammatory bowel disease impacts diagnosis. Scientific Reports 2020, 10, 1-11, 10.1038/s41598-020-71503-y.
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760–1765. Saul A. Villeda; Jian Luo; Kira Mosher; Bende Zou; Markus Britschgi; Gregor Bieri; Trisha M. Stan; Nina Fainberg; Zhaoqing Ding; Alexander Eggel; et al.Kurt M. LucinEva CzirrJeong-Soo ParkSebastien Couillard-DespresLudwig AignerGe LiElaine R. PeskindJeffrey KayeJoseph QuinnDouglas R. GalaskoXinmin Simon XieThomas RandoTony Wyss-Coray The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90-94, 10.1038/nature10357.
- Korolkova, O.Y.; Myers, J.N.; Pellom, S.T.; Wang, L.; M’Koma, A.E. Characterization of Serum Cytokine Profile in Predominantly Colonic Inflammatory Bowel Disease to Delineate Ulcerative and Crohn’s Colitides. Clin. Med. Insights Gastroenterol. 2015, 8, 29–44. Ioannis-Alexandros Gampierakis; Yassemi Koutmani; Maria Semitekolou; Ioannis Morianos; Alexia Polissidis; Antonia Katsouda; Ioannis Charalampopoulos; Georgina Xanthou; Achille Gravanis; Katia P. Karalis; et al. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Molecular Psychiatry 2020, 26, 1248-1263, 10.1038/s41380-020-0651-6.
- Tatsuki, M.; Hatori, R.; Nakazawa, T.; Ishige, T.; Hara, T.; Kagimoto, S.; Tomomasa, T.; Arakawa, H.; Takizawa, T. Serological cytokine signature in paediatric patients with inflammatory bowel disease impacts diagnosis. Sci. Rep. 2020, 10, 14638. Eloisa Salvo; Patricia Stokes; Ciara E. Keogh; Ingrid Brust-Mascher; Carly Hennessey; Trina A. Knotts; Jessica A. Sladek; Kavi M. Rude; Michelle Swedek; Gonzalo Rabasa; et al.Mélanie G. Gareau A murine model of pediatric inflammatory bowel disease causes microbiota-gut-brain axis deficits in adulthood. American Journal of Physiology-Gastrointestinal and Liver Physiology 2020, 319, G361-G374, 10.1152/ajpgi.00177.2020.
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. Svetlana Zonis; Robert N Pechnick; Vladimir A Ljubimov; Michael Mahgerefteh; Kolja Wawrowsky; Kathrin S Michelsen; Vera Chesnokova; Chronic intestinal inflammation alters hippocampal neurogenesis. Journal of Neuroinflammation 2015, 12, 1-12, 10.1186/s12974-015-0281-0.
- Gampierakis, I.-A.; Koutmani, Y.; Semitekolou, M.; Morianos, I.; Polissidis, A.; Katsouda, A.; Charalampopoulos, I.; Xanthou, G.; Gravanis, A.; Karalis, K.P. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Mol. Psychiatry 2021, 26, 1248–1263. Osamu Nakagawasai; Kotaro Yamada; Kohei Takahashi; Takayo Odaira; Wakana Sakuma; Daisuke Ishizawa; Naruya Takahashi; Kentaro Onuma; Chikako Hozumi; Wataru Nemoto; et al.Koichi Tan-No Liver hydrolysate prevents depressive-like behavior in an animal model of colitis: Involvement of hippocampal neurogenesis via the AMPK/BDNF pathway. Behavioural Brain Research 2020, 390, 112640, 10.1016/j.bbr.2020.112640.
- Salvo, E.; Stokes, P.; Keogh, C.E.; Brust-Mascher, I.; Hennessey, C.; Knotts, T.A.; Sladek, J.A.; Rude, K.M.; Swedek, M.; Rabasa, G.; et al. A murine model of pediatric inflammatory bowel disease causes microbiota-gut-brain axis deficits in adulthood. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G361–G374. Kohei Takahashi; Kazuhiro Kurokawa; Kazuya Miyagawa; Atsumi Mochida-Saito; Yukio Nemoto; Hiroyuki Iwasa; Osamu Nakagawasai; Takeshi Tadano; Hiroshi Takeda; Minoru Tsuji; et al. Antidementia effects of Enterococcus faecalis 2001 are associated with enhancement of hippocampal neurogenesis via the ERK-CREB-BDNF pathway in olfactory bulbectomized mice. Physiology & Behavior 2020, 223, 112997, 10.1016/j.physbeh.2020.112997.
- Zonis, S.; Pechnick, R.N.; Ljubimov, V.A.; Mahgerefteh, M.; Wawrowsky, K.; Michelsen, K.S.; Chesnokova, V. Chronic intestinal inflammation alters hippocampal neurogenesis. J. Neuroinflamm. 2015, 12, 65. Hyungju Park; Mu-Ming Poo; Neurotrophin regulation of neural circuit development and function. Nature Reviews Neuroscience 2012, 14, 7-23, 10.1038/nrn3379.
- Nakagawasai, O.; Yamada, K.; Takahashi, K.; Odaira, T.; Sakuma, W.; Ishizawa, D.; Takahashi, N.; Onuma, K.; Hozumi, C.; Nemoto, W.; et al. Liver hydrolysate prevents depressive-like behavior in an animal model of colitis: Involvement of hippocampal neurogenesis via the AMPK/BDNF pathway. Behav. Brain Res. 2020, 390, 112640. Plinio C. Casarotto; Mykhailo Girych; Senem M. Fred; Vera Kovaleva; Rafael Moliner; Giray Enkavi; Caroline Biojone; Cecilia Cannarozzo; Madhusmita Pryiadrashini Sahu; Katja Kaurinkoski; et al.Cecilia A. BrunelloAnna SteinzeigFrederike WinkelSudarshan PatilStefan VestringTsvetan SerchovCassiano R.A.F. DinizLiina LaukkanenIseline CardonHanna AntilaTomasz RogTimo Petteri PiepponenClive R. BramhamClaus NormannSari E. LauriMart SaarmaIlpo VattulainenEero Castrén Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299-1313.e19, 10.1016/j.cell.2021.01.034.
- Takahashi, K.; Kurokawa, K.; Miyagawa, K.; Mochida-Saito, A.; Nemoto, Y.; Iwasa, H.; Nakagawasai, O.; Tadano, T.; Takeda, H.; Tsuji, M. Antidementia effects of Enterococcus faecalis 2001 are associated with enhancement of hippocampal neurogenesis via the ERK-CREB-BDNF pathway in olfactory bulbectomized mice. Physiol. Behav. 2020, 223, 112997. R.M Barrientos; D.B Sprunger; S Campeau; E.A Higgins; L.R Watkins; J.W Rudy; S.F Maier; Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847-853, 10.1016/s0306-4522(03)00564-5.
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. Xiao Zhuang; Bing Zhan; Yufeng Jia; Chaoze Li; Nan Wu; Ming Zhao; Nuo Chen; Yaxin Guo; Yingxin Du; Yi Zhang; et al.Baihui CaoYan LiFaliang ZhuChun GuoQun WangLining Zhang IL-33 in the basolateral amygdala integrates neuroinflammation into anxiogenic circuits via modulating BDNF expression. Brain, Behavior, and Immunity 2022, 102, 98-109, 10.1016/j.bbi.2022.02.019.
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19. Arya Haj-Mirzaian; Shayan Amiri; Hossein Amini-Khoei; Mir-Jamal Hosseini; Arvin Haj-Mirzaian; Majid Momeny; Maryam Rahimi-Balaei; Ahmad Reza Dehpour; Anxiety- and Depressive-Like Behaviors are Associated with Altered Hippocampal Energy and Inflammatory Status in a Mouse Model of Crohn’s Disease. Neuroscience 2017, 366, 124-137, 10.1016/j.neuroscience.2017.10.023.
- Barrientos, R.; Sprunger, D.; Campeau, S.; Higgins, E.; Watkins, L.; Rudy, J.; Maier, S. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847–853. Kiarash Riazi; Michael A. Galic; J. Brent Kuzmiski; Winnie Ho; Keith A. Sharkey; Quentin J. Pittman; Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proceedings of the National Academy of Sciences 2008, 105, 17151-17156, 10.1073/pnas.0806682105.
- Zhuang, X.; Zhan, B.; Jia, Y.; Li, C.; Wu, N.; Zhao, M.; Chen, N.; Guo, Y.; Du, Y.; Zhang, Y.; et al. IL-33 in the basolateral amygdala integrates neuroinflammation into anxiogenic circuits via modulating BDNF expression. Brain Behav. Immun. 2022, 102, 98–109. Xiaomin Yuan; Biqing Chen; Zhenglan Duan; Ziqian Xia; Yang Ding; Tuo Chen; Huize Liu; Baosheng Wang; Bolin Yang; Xiaoyong Wang; et al.Shijia LiuJin-Yong ZhouYajun LiuQiong WangZhaofeng ShenJun XiaoHongtao ShangWeiwei LiuGuoping ShiLei ZhuYugen Chen Depression and anxiety in patients with active ulcerative colitis: crosstalk of gut microbiota, metabolomics and proteomics. Gut Microbes 2021, 13, 1987779, 10.1080/19490976.2021.1987779.
- Haj-Mirzaian, A.; Amiri, S.; Amini-Khoei, H.; Hosseini, M.-J.; Haj-Mirzaian, A.; Momeny, M.; Rahimi-Balaei, M.; Dehpour, A.R. Anxiety- and Depressive-Like Behaviors are Associated with Altered Hippocampal Energy and Inflammatory Status in a Mouse Model of Crohn’s Disease. Neuroscience 2017, 366, 124–137. Fernando A. Vicentini; Jake C. Szamosi; Laura Rossi; Lateece Griffin; Kristoff Nieves; Dominique Bihan; Ian A. Lewis; Quentin J. Pittman; Mark G. Swain; Michael G. Surette; et al.Simon A. HirotaKeith A. Sharkey Colitis-associated microbiota drives changes in behaviour in male mice in the absence of inflammation. Brain, Behavior, and Immunity 2022, 102, 266-278, 10.1016/j.bbi.2022.03.001.
- Heydarpour, P.; Rahimian, R.; Fakhfouri, G.; Khoshkish, S.; Fakhraei, N.; Salehi-Sadaghiani, M.; Wang, H.; Abbasi, A.; Dehpour, A.R.; Ghia, J.E. Behavioral despair associated with a mouse model of Crohn’s disease: Role of nitric oxide pathway. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 131–141. Mireia Valles-Colomer; Gwen Falony; Youssef Darzi; Ettje F. Tigchelaar; Jun Wang; Raul Y. Tito; Carmen Schiweck; Alexander Kurilshikov; Marie Joossens; Cisca Wijmenga; et al.Stephan ClaesLukas Van OudenhoveAlexandra ZhernakovaSara Vieira-SilvaJeroen Raes The neuroactive potential of the human gut microbiota in quality of life and depression. Nature Microbiology 2019, 4, 623-632, 10.1038/s41564-018-0337-x.
- Yuan, X.; Chen, B.; Duan, Z.; Xia, Z.; Ding, Y.; Chen, T.; Liu, H.; Wang, B.; Yang, B.; Wang, X.; et al. Depression and anxiety in patients with active ulcerative colitis: Crosstalk of gut microbiota, metabolomics and proteomics. Gut Microbes 2021, 13, 1987779. Kohei Takahashi; Osamu Nakagawasai; Wataru Nemoto; Takayo Odaira; Wakana Sakuma; Hiroshi Onogi; Hiroaki Nishijima; Ryuji Furihata; Yukio Nemoto; Hiroyuki Iwasa; et al.Koichi Tan-NoTakeshi Tadano Effect of Enterococcus faecalis 2001 on colitis and depressive-like behavior in dextran sulfate sodium-treated mice: involvement of the brain–gut axis. Journal of Neuroinflammation 2019, 16, 1-16, 10.1186/s12974-019-1580-7.
- Vicentini, F.A.; Szamosi, J.C.; Rossi, L.; Griffin, L.; Nieves, K.; Bihan, D.; Lewis, I.A.; Pittman, Q.J.; Swain, M.G.; Surette, M.G.; et al. Colitis-associated microbiota drives changes in behaviour in male mice in the absence of inflammation. Brain Behav. Immun. 2022, 102, 266–278. Jacob R. Emge; Kevin Huynh; Elaine N. Miller; Manvir Kaur; Colin Reardon; Kim E. Barrett; Mélanie G. Gareau; Modulation of the microbiota-gut-brain axis by probiotics in a murine model of inflammatory bowel disease. American Journal of Physiology-Gastrointestinal and Liver Physiology 2016, 310, G989-G998, 10.1152/ajpgi.00086.2016.
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. Roman Sankowski; Jasmin Ahmari; Charlotte Mezö; Anna Lena Hrabě de Angelis; Vidmante Fuchs; Olaf Utermöhlen; Thorsten Buch; Thomas Blank; Mercedes Gomez de Agüero; Andrew J Macpherson; et al.Daniel Erny Commensal microbiota divergently affect myeloid subsets in the mammalian central nervous system during homeostasis and disease. The EMBO Journal 2021, 40, e108605, 10.15252/embj.2021108605.
- Zhang, Y.; Fan, Q.; Hou, Y.; Zhang, X.; Yin, Z.; Cai, X.; Wei, W.; Wang, J.; He, D.; Wang, G.; et al. Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain Behav. Immun. 2022, 102, 11–22. Alba Rodríguez-Nogales; Francesca Algieri; José Garrido-Mesa; Teresa Vezza; M. Pilar Utrilla; Natalia Chueca; Federico García; M. Elena Rodríguez-Cabezas; Julio Gálvez; Intestinal anti-inflammatory effect of the probiotic Saccharomyces boulardii in DSS-induced colitis in mice: Impact on microRNAs expression and gut microbiota composition. The Journal of Nutritional Biochemistry 2018, 61, 129-139, 10.1016/j.jnutbio.2018.08.005.
- Takahashi, K.; Nakagawasai, O.; Nemoto, W.; Odaira, T.; Sakuma, W.; Onogi, H.; Nishijima, H.; Furihata, R.; Nemoto, Y.; Iwasa, H.; et al. Effect of Enterococcus faecalis 2001 on colitis and depressive-like behavior in dextran sulfate sodium-treated mice: Involvement of the brain–gut axis. J. Neuroinflamm. 2019, 16, 201. Daniel Erny; Nikolaos Dokalis; Charlotte Mezö; Angela Castoldi; Omar Mossad; Ori Staszewski; Maximilian Frosch; Matteo Villa; Vidmante Fuchs; Arun Mayer; et al.Jana NeuberJanika SosatStefan TholenOliver SchillingAndreas VlachosThomas BlankMercedes Gomez de AgüeroAndrew J. MacphersonEdward J. PearceMarco Prinz Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metabolism 2021, 33, 2260-2276.e7, 10.1016/j.cmet.2021.10.010.
- Emge, J.R.; Huynh, K.; Miller, E.N.; Kaur, M.; Reardon, C.; Barrett, K.E.; Gareau, M.G. Modulation of the microbiota-gut-brain axis by probiotics in a murine model of inflammatory bowel disease. Am. J. Physiol. Liver Physiol. 2016, 310, G989–G998. Daniel Erny; Anna Lena Hrabě De Angelis; Diego Adhemar Jaitin; Peter Wieghofer; Ori Staszewski; Eyal David; Hadas Keren-Shaul; Tanel Mahlakoiv; Kristin Jakobshagen; Thorsten Buch; et al.Vera SchwierzeckOlaf UtermöhlenEunyoung ChunWendy GarrettKathy D McCoyAndreas DiefenbachPeter StaeheliBärbel StecherIdo AmitMarco Prinz Host microbiota constantly control maturation and function of microglia in the CNS. Nature Neuroscience 2015, 18, 965-977, 10.1038/nn.4030.
- Sankowski, R.; Ahmari, J.; Mezö, C.; de Angelis, A.L.H.; Fuchs, V.; Utermöhlen, O.; Buch, T.; Blank, T.; de Agüero, M.G.; Macpherson, A.J.; et al. Commensal microbiota divergently affect myeloid subsets in the mammalian central nervous system during homeostasis and disease. EMBO J. 2021, 40, e108605. Omar Mossad; Bérénice Batut; Bahtiyar Yilmaz; Nikolaos Dokalis; Charlotte Mezö; Elisa Nent; Lara Susann Nabavi; Melanie Mayer; Feres José Mocayar Maron; Joerg M. Buescher; et al.Mercedes Gomez de AgüeroAntal SzalayTim LämmermannAndrew J. MacphersonStephanie C. Ganal-VonarburgRolf BackofenDaniel ErnyMarco PrinzThomas Blank Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N6-carboxymethyllysine. Nature Neuroscience 2022, 25, 295-305, 10.1038/s41593-022-01027-3.
- Rodríguez-Nogales, A.; Algieri, F.; Garrido-Mesa, J.; Vezza, T.; Utrilla, M.P.; Chueca, N.; García, F.; Rodríguez-Cabezas, M.E.; Gálvez, J. Intestinal anti-inflammatory effect of the probiotic Saccharomyces boulardii in DSS-induced colitis in mice: Impact on microRNAs expression and gut microbiota composition. J. Nutr. Biochem. 2018, 61, 129–139. Alexander Duscha; Barbara Gisevius; Sarah Hirschberg; Nissan Yissachar; Gabriele I. Stangl; Eva Eilers; Verian Bader; Stefanie Haase; Johannes Kaisler; Christina David; et al.Ruth SchneiderRiccardo TroisiDaniel ZentTobias HegelmaierNikolaos DokalisSara GersteinSara Del Mare-RoumaniSivan AmidrorOri StaszewskiGereon PoschmannKai StühlerFrank HircheAndras BaloghStefan KempaPascal TrägerMario M. ZaissJacob Bak HolmMegan G. MassaHenrik Bjørn NielsenAndreas FaissnerCarsten LukasSören G. GatermannMarkus ScholzHorst PrzuntekMarco PrinzSofia K. ForslundKonstanze F. WinklhoferDominik N. MüllerRalf A. LinkerRalf GoldAiden Haghikia Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067-1080.e16, 10.1016/j.cell.2020.02.035.
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7. Veit Rothhammer; Davis M. Borucki; Emily C. Tjon; Maisa C. Takenaka; Chun-Cheih Chao; Alberto Ardura-Fabregat; Kalil Alves de Lima; Cristina Gutiérrez-Vázquez; Patrick Hewson; Ori Staszewski; et al.Manon BlainLuke HealyTradite NezirajMatilde BorioMichael WheelerLoic Lionel DraginDavid LaplaudJack AntelJorge Ivan AlvarezMarco PrinzFrancisco J. Quintana Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724-728, 10.1038/s41586-018-0119-x.
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. Irina Leonardi; Iris H. Gao; Woan-Yu Lin; Megan Allen; Xin V. Li; William D. Fiers; Meghan Bialt De Celie; Gregory G. Putzel; Rhonda K. Yantiss; Melanie Johncilla; et al.Dilek ColakIliyan D. Iliev Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 2022, 185, 831-846.e14, 10.1016/j.cell.2022.01.017.
- Mossad, O.; Batut, B.; Yilmaz, B.; Dokalis, N.; Mezö, C.; Nent, E.; Nabavi, L.S.; Mayer, M.; Maron, F.J.M.; Buescher, J.M.; et al. Gut microbiota drives age-related oxidative stress and mitochondrial damage in microglia via the metabolite N6-carboxymethyllysine. Nat. Neurosci. 2022, 25, 295–305. Miriam Bittel; Patrick Reichert; Ilann Sarfati; Anja Dressel; Stefanie Leikam; Stefan Uderhardt; Iris Stolzer; Tuan Anh Phu; Martin Ng; Ngan K. Vu; et al.Stefan TenzerUte DistlerStefan WirtzVeit RothhammerMarkus F. NeurathRobert L. RaffaiClaudia GüntherStefan Momma Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe‐host‐communication in vivo. Journal of Extracellular Vesicles 2021, 10, e12159, 10.1002/jev2.12159.
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. Youying Zhang; Qilin Fan; Yuanlong Hou; Xuanshuang Zhang; Zhe Yin; Xiaoying Cai; Wei Wei; Jiaying Wang; Dandan He; Guangji Wang; et al.Yonggui YuanHaiping HaoXiao Zheng Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain, Behavior, and Immunity 2022, 102, 11-22, 10.1016/j.bbi.2022.02.007.
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. Seth M. Bloom; Vinieth N. Bijanki; Gerardo M. Nava; Lulu Sun; Nicole P. Malvin; David L. Donermeyer; W. Michael Dunne; Paul M. Allen; Thaddeus S. Stappenbeck; Commensal Bacteroides Species Induce Colitis in Host-Genotype-Specific Fashion in a Mouse Model of Inflammatory Bowel Disease. Cell Host & Microbe 2011, 9, 390-403, 10.1016/j.chom.2011.04.009.
- Leonardi, I.; Gao, I.H.; Lin, W.-Y.; Allen, M.; Li, X.V.; Fiers, W.D.; De Celie, M.B.; Putzel, G.G.; Yantiss, R.K.; Johncilla, M.; et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 2022, 185, 831–846.e14. Leon M. T. Dicks; Diron Hurn; Demi Hermanus; Gut Bacteria and Neuropsychiatric Disorders. Microorganisms 2021, 9, 2583, 10.3390/microorganisms9122583.
- Bittel, M.; Reichert, P.; Sarfati, I.; Dressel, A.; Leikam, S.; Uderhardt, S.; Stolzer, I.; Phu, T.A.; Ng, M.; Vu, N.K.; et al. Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J. Extracell. Vesicles 2021, 10, e12159. Jian-Jun Chen; Ben-Hua Zeng; Wen-Wen Li; Chan-Juan Zhou; Song-Hua Fan; Ke Cheng; Li Zeng; Peng Zheng; Liang Fang; Hong Wei; et al.Peng Xie Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behavioural Brain Research 2017, 322, 34-41, 10.1016/j.bbr.2017.01.021.
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides Species Induce Colitis in Host-Genotype-Specific Fashion in a Mouse Model of Inflammatory Bowel Disease. Cell Host Microbe 2011, 9, 390–403. A E Hoban; R M Stilling; Gerard Moloney; F Shanahan; T G Dinan; G Clarke; J F Cryan; The microbiome regulates amygdala-dependent fear recall. Molecular Psychiatry 2017, 23, 1134-1144, 10.1038/mp.2017.100.
- Dicks, L.M.T.; Hurn, D.; Hermanus, D. Gut Bacteria and Neuropsychiatric Disorders. Microorganisms 2021, 9, 2583. Coco Chu; Mitchell H. Murdock; Deqiang Jing; Tae Hyung Won; Hattie Chung; Adam Kressel; Tea Tsaava; Meghan E. Addorisio; Gregory G. Putzel; Lei Zhou; et al.Nicholas J. BessmanRuirong YangSaya MoriyamaChristopher N. ParkhurstAnfei LiHeidi C. MeyerFei TengSangeeta S. ChavanKevin J TraceyAviv RegevFrank C. SchroederFrancis S. LeeConor ListonDavid Artis The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543-548, 10.1038/s41586-019-1644-y.
- Chen, J.-J.; Zeng, B.-H.; Li, W.-W.; Zhou, C.-J.; Fan, S.-H.; Cheng, K.; Zeng, L.; Zheng, P.; Fang, L.; Wei, H.; et al. Effects of gut microbiota on the microRNA and mRNA expression in the hippocampus of mice. Behav. Brain Res. 2017, 322, 34–41. Omar Mossad; Elisa Nent; Sabrina Woltemate; Shani Folschweiller; Joerg M. Buescher; Daniel Schnepf; Daniel Erny; Peter Staeheli; Marlene Bartos; Antal Szalay; et al.Bärbel StecherMarius VitalJonas F. SauerTim LämmermannMarco PrinzThomas Blank Microbiota-dependent increase in δ-valerobetaine alters neuronal function and is responsible for age-related cognitive decline. Nature Aging 2021, 1, 1127-1136, 10.1038/s43587-021-00141-4.
- Hoban, A.E.; Stilling, R.M.; Moloney, G.; Shanahan, F.; Dinan, T.G.; Clarke, G.; Cryan, J.F. The microbiome regulates amygdala-dependent fear recall. Mol. Psychiatry 2018, 23, 1134–1144. Fernando A. Vicentini; Catherine M. Keenan; Laurie E. Wallace; Crystal Woods; Jean-Baptiste Cavin; Amanda R. Flockton; Wendy B. Macklin; Jaime Belkind-Gerson; Simon A. Hirota; Keith A. Sharkey; et al. Intestinal microbiota shapes gut physiology and regulates enteric neurons and glia. Microbiome 2021, 9, 1-24, 10.1186/s40168-021-01165-z.
- Casado-Bedmar, M.; Viennois, E. MicroRNA and Gut Microbiota: Tiny but Mighty—Novel Insights into Their Cross-talk in Inflammatory Bowel Disease Pathogenesis and Therapeutics. J. Crohn’s Colitis 2021, 16, 992–1005.
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L.; et al. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548.
- Mossad, O.; Nent, E.; Woltemate, S.; Folschweiller, S.; Buescher, J.M.; Schnepf, D.; Erny, D.; Staeheli, P.; Bartos, M.; Szalay, A.; et al. Microbiota-dependent increase in δ-valerobetaine alters neuronal function and is responsible for age-related cognitive decline. Nat. Aging 2021, 1, 1127–1136.
- Vicentini, F.A.; Keenan, C.M.; Wallace, L.E.; Woods, C.; Cavin, J.-B.; Flockton, A.R.; Macklin, W.B.; Belkind-Gerson, J.; Hirota, S.A.; Sharkey, K.A. Intestinal microbiota shapes gut physiology and regulates enteric neurons and glia. Microbiome 2021, 9, 210.