 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Junjie Luo | -- | 1997 | 2022-06-23 06:23:57 | | | |
| 2 | Junjie Luo | -6 word(s) | 1991 | 2022-06-23 06:40:08 | | | | |
| 3 | Junjie Luo | Meta information modification | 1991 | 2022-06-23 06:50:13 | | | | |
| 4 | Conner Chen | Meta information modification | 1991 | 2022-06-23 07:49:25 | | |
Video Upload Options
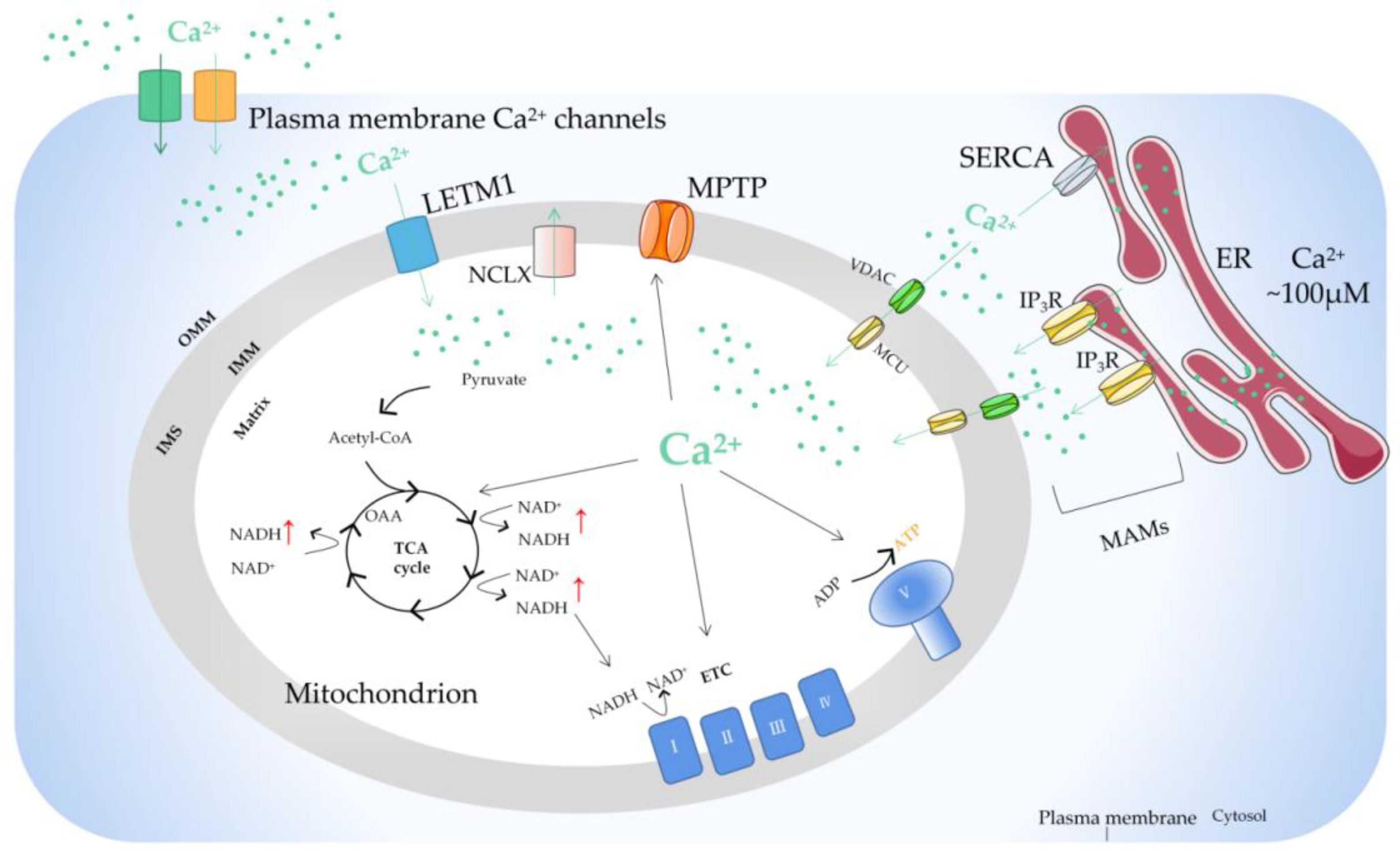
Mitochondria, as the main site of cellular energy metabolism and the generation of oxygen free radicals, are the key switch for mitochondria-mediated endogenous apoptosis. Ca2+ is not only an important messenger for cell proliferation, but it is also an indispensable signal for cell death. Ca2+ participates in and plays a crucial role in the energy metabolism, physiology, and pathology of mitochondria. Mitochondria control the uptake and release of Ca2+ through channels/transporters, such as the mitochondrial calcium uniporter (MCU), and influence the concentration of Ca2+ in both mitochondria and cytoplasm, thereby regulating cellular Ca2+ homeostasis. Mitochondrial Ca2+ transport-related processes are involved in important biological processes of tumor cells including proliferation, metabolism, and apoptosis.
1. Introduction
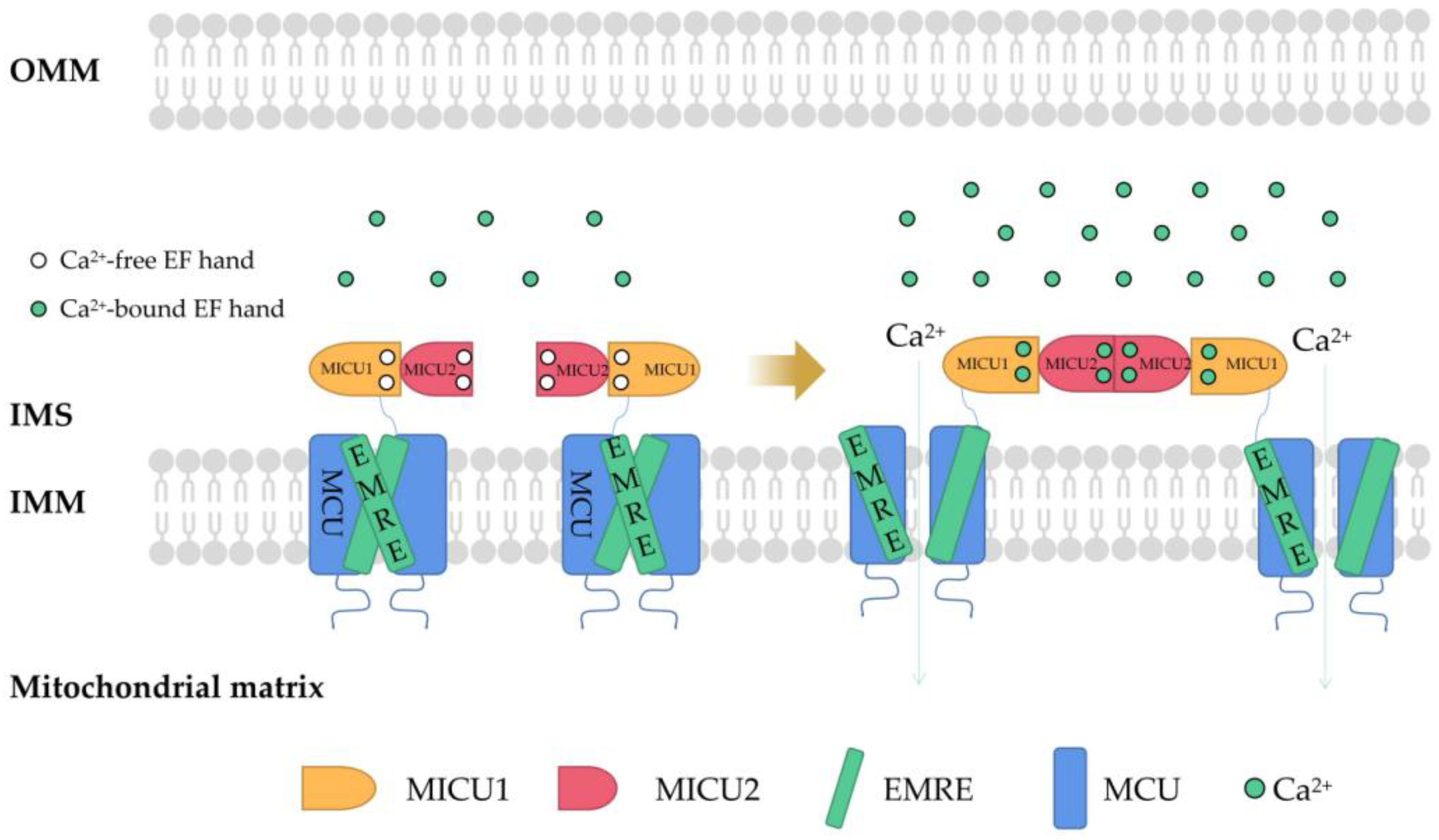
2. Regulation of Mitochondrial Ca2+
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Chem. Commun. 2012, 148, 1145–1159.
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634.
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323.
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730.
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168.
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. JBC 2016, 40, 20849–20857.
- Zavodnik, I.B. Mitochondria, calcium homeostasis and calcium signaling. Biomed. Khim. 2016, 62, 311–317.
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565.
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018, 69, 62–72.
- Magalhães, P.J.; Rizzuto, R. Mitochondria and calcium homeostasis: A tale of three luminescent proteins. Luminescence 2001, 16, 67–71.
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012.
- Robb-Gaspers, L.; Burnett, P.; Rutter, G.; Denton, R.; Rizzuto, R.; Thomas, A. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000.
- Uhlén, P.; Fritz, N. Biochemistry of calcium oscillations. Biochem. Biophys. Res. Commun. 2010, 396, 28–32.
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747.
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424.
- Sheu, S.S.; Jou, M.J. Mitochondrial free Ca2+ concentration in living cells. J. Bioenerg. Biomembr. 1994, 26, 487–493.
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812.
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529.
- Paudel, S.; Sindelar, R.; Saha, M. Calcium Signaling in Vertebrate Development and Its Role in Disease. Int. J. Mol. Sci. 2018, 19, 3390.
- Pézier, A.; Acquistapace, A.; Renou, M.; Rospars, J.P.; Lucas, P. Ca2+ stabilizes the membrane potential of moth olfactory receptor neurons at rest and is essential for their fast repolarization Chem. Senses 2007, 32, 305–317.
- Pendin, D.; Greotti, E.; Filadi, R.; Pozzan, T. Spying on organelle Ca2+ in living cells: The mitochondrial point of view. J. Endocrinol. Investig. 2015, 38, 39–45.
- Yamamoto, T. The Molecular Mechanisms of Mitochondrial Calcium Uptake by Calcium Uniporter. Yakugaku Zasshi J. Pharm. Jpn. 2021, 141, 491–499.
- Stefani, D.D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340.
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345.
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472.
- Garbincius, J.; Elrod, J. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992.
- Chen, L.; Sun, Q.; Zhou, D.; Song, W.; Yang, Q.; Ju, B.; Zhang, L.; Xie, H.; Zhou, L.; Hu, Z.; et al. HINT2 triggers mitochondrial Ca2+ influx by regulating the mitochondrial Ca2+ uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017, 411, 106–116.
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700.
- Sancak, Y.; Markhard, A.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.; Udeshi, N.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382.
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58.
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell. 2020, 12, 220–229.
- Fan, M.; Zhang, J.; Tsai, C.; Benjamin, J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133.
- Tomar, D.; Elrod, J.W. Metabolite regulation of the mitochondrial calcium uniporter channel. Cell Calcium. 2020, 92, 102288.
- Gottschalk, B.; Madreiter-Sokolowski, C.T.; Graier, W.F. Cristae junction as a fundamental switchboard for mitochondrial ion signaling and bioenergetics. Cell Calcium. 2022, 101, 102517.
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732.
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634.
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452.
- Madreiter-Sokolowski, C.T.; Klec, C.; Parichatikanond, W.; Stryeck, S.; Gottschalk, B.; Pulido, S.; Rost, R.; Eroglu, E.; Hofmann, N.A.; Bondarenko, A.I.; et al. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca(2+) uptake in immortalized cells. Nat. Commun. 2016, 7, 12897.
- Madreiter-Sokolowski, C.T.; Győrffy, B.; Klec, C.; Sokolowski, A.A.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 and PRMT1 are key prognostic markers for lung carcinoma patients. Oncotarget. 2017, 8, 80278–80285.
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009.
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J. MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Reports. 2017, 21, 3141–3154.
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195.
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376.
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.; Thomas, T.; Hoffman, N.; Timbalia, S.; Goldman, S.; Breves, S.; Corbally, D.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685.
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880.
- Roy, S.; Dey, K.; Hershfinkel, M.; Ohana, E.; Sekler, I. Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 997–1008.
- Starkov, A.A.; Chinopoulos, C.; Starkova, N.N.; Konrad, C.; Kiss, G.; Stepanova, A.; Popov, V.N. Divalent cation chelators citrate and EDTA unmask an intrinsic uncoupling pathway in isolated mitochondria. J. Bioenerg. Biomembr. 2017, 49, 3–11.
- Wilson, J.A.; Lau, Y.S.; Gleeson, J.G.; Wilson, J.S. The action of MPTP on synaptic transmission is affected by changes in Ca2+ concentrations. Brain Res. 1991, 541, 342–346.
- Kolomiiets’, O.; Danylovych, I.; Danylovych, H. H+-Ca2+-exchanger in the myometrium mitochondria: Modulation of exogenous and endogenous compounds. Fiziol. Zh. 2014, 60, 33–42.
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385.
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784.
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534.
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340.
- Glitsch, M. Mechano- and pH-sensing convergence on Ca2+-mobilising proteins - A recipe for cancer? Cell Calcium. 2019, 80, 38–45.
- Trapani, V.; Wolf, F.I. Dysregulation of Mg2+ homeostasis contributes to acquisition of cancer hallmarks. Cell Calcium. 2019, 83, 102078.
- Santoni, G.; Morelli, M.B.; Marinelli, O.; Nabissi, M.; Santoni, M.; Amantini, C. Calcium Signaling and the Regulation of Chemosensitivity in Cancer Cells: Role of the Transient Receptor Potential Channels. Adv. Exp. Med. Biol. 2020, 1131, 505–517.


