Mitochondria are involved in a series of cellular biological processes such as adenosine triphosphate (ATP) generation, apoptosis, and cell cycle regulation to maintain the cell’s life activities [
1]. Calcium ions (Ca
2+) are distributed in the mitochondrial intermembrane gap and matrix [
2]. Ca
2+ shuttles between mitochondria and cytoplasm through different transport mechanisms, regulating the life activities of mitochondria and even the whole cell. Ca
2+ is an indispensable messenger for many important physiologic processes, including metabolism, cell proliferation and death, protein phosphorylation, gene transcription, neurotransmission, contraction, and secretion [
3]. The level of intracellular Ca
2+ depends on the release of endoplasmic reticulum (ER) Ca
2+ and the inflow of extracellular Ca
2+ [
4]. In animal body fluids and tissues, the concentration of Ca
2+ varies between 2.1 and 2.6 mM [
5] and the unit of total Ca
2+ concentration in cells is also mM. However, in the cytoplasm of most cells, the concentration of free Ca
2+ is about 10,000 times lower. In cells, inorganic compounds and low molecular weight organic molecules usually bind Ca
2+ with low affinity and will not reduce their free concentration to nM, which is necessary for Ca
2+ to effectively perform their signaling functions [
6]. Abnormal Ca
2+ homeostasis is one of the common pathological mechanisms of many diseases. Studies have shown that Ca
2+ can not only be absorbed and released by mitochondria, but also the process of Ca
2+ uptake and release by mitochondria plays an important role in maintaining cytoplasmic calcium homeostasis [
7,
8,
9,
10,
11].
Ca
2+ plays an indispensable role in signal transduction from cell surface receptors to the cytoplasm and from the cytoplasm to mitochondria, so as to jointly regulate cell metabolism [
12]. Cytoplasmic calcium oscillation is the most prominent signal in cells, which refers to the transmission of a variety of regulatory information by cytosolic Ca
2+ ([Ca
2+]c) in the form of concentration oscillation [
13]. Inositol 1,4,5-trisphosphate (IP
3)-induced intracellular Ca
2+ mobilization results in an increase in mitochondrial Ca
2+ ([Ca
2+]m) [
14]. IP
3-dependent hormone-induced [Ca
2+]c oscillation is effectively transmitted to mitochondria in the form of [Ca
2+]m oscillation [
15]. Moreover, it has been reported that the concentration of free Ca
2+ in mitochondria is closely related to the level of energy metabolism and the change in membrane permeability [
16]. Mitochondrial Ca
2+ accumulation triggers the activation of the mitochondrial metabolic mechanism, which increases ATP synthesis in the mitochondria and the ATP level in cytoplasm [
17]. The uptake and release of mitochondrial Ca
2+ also affects the intracellular calcium signal [
18]. The abnormality in these calcium signaling-related activities is significantly related to the occurrence and development of heart disease, epilepsy, and neurodegenerative diseases [
19].
2. Regulation of Mitochondrial Ca2+
Due to the outer membrane of mitochondria possessing a high permeability for Ca
2+, the concentration of Ca
2+ in the membrane gap is equivalent to that in the cytoplasm [
35]. In the resting state of cells, the concentration of Ca
2+ in cytoplasm is about 100nM. When the cells are excited, the concentration of Ca
2+ in the cytoplasm can rise to 1–3 µM. [
36]. In fact, the uptake and release of Ca
2+ by mitochondria can be regulated by the one-way transport mechanism or transporter [
37]. The mitochondrial Ca
2+ influx is mainly mediated by the mitochondrial calcium uniporter (MCU), voltage-dependent anion-selective channel (VDAC), and mitochondrial ryanodine receptor transporter. Furthermore, the mitochondrial Ca
2+ efflux pathways mainly include leucine zipper/EF hand-containing transmembrane-1 (LETM1), mitochondrial Na
+/Ca
2+ exchanger (NCLX), and mitochondrial permeability transition pore (MPTP).
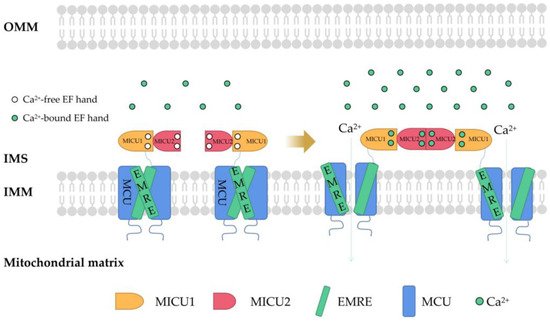
MCU is a Ca
2+ channel ubiquitous in mitochondrial intima [
38]. It is generally considered to be a key Ca
2+ transporter [
39] and silencing the MCU can severely abrogate mitochondrial Ca
2+ uptake [
40] (
Figure 1). Knockout of the MCU completely inhibited mitochondrial Ca
2+ uptake triggered by several stimuli in different cell types [
41]. The MCU is an ion channel with electrophysiological characteristics. Ca
2+ uptake through the MCU is driven by an electrochemical gradient. The MCU and related regulating molecules, including the essential MCU regulator (EMRE), mitochondrial calcium uptake (MICU)1, MICU2, MICU3, MCU-dominant negative beta subunit (MCUb), and MCU regulator 1 (MCUR1), form a large complex to manipulate the activities of the MCU [
42]. The changes in the expression of these regulators are different in different cancer cells. For example, in pancreatic cancer cells, MICU1 and MICU2 are increased, while EMRE is decreased [
43]. In breast cancer cells, MCU is elevated but MCUb is reduced [
44]. In ovarian cancer cells, MICU1 mRNA is enhanced [
45]. In HCC cells, the MCU, MCUR1, and MICU2 are elevated, while MICU1 is in decline [
46].
Figure 1. The structure of MCU and connections to its regulators. Mitochondrial Ca2+ uptake through MCU. In mammals, MCU contains four core components: pore-forming MCU protein, the gatekeepers MICU1 and MICU2, and an auxiliary subunit EMRE. MCU plays a vital role in Ca2+ transport. In order to prevent Ca2+ overload, the activity of MCU must be strictly regulated by MICUs, which can sense the change in cytosolic Ca2+ concentration to open and close the MCU. MCU, mitochondrial calcium uniporter; MICU, mitochondrial Ca2+ uptake; EMRE, essential MCU regulator; OMM, outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane; EF hand, helix−loop−helix structure.
In higher eukaryotes, the EMRE mediates MICU1/MICU2 to regulate Ca
2+ transport through a leverage mechanism. MICU1/MICU2 is associated with the MCU through the EMRE. Each MICU1 interacts with two EMRE subunits. The interaction sites are located at the N-terminal poly K, s339k340k341 domain of MICU1 and the C-terminal of the EMRE [
47]. The regulation of MCU activity by MICU1 and MICU2 involves a gating mechanism: when cells in a resting state and the concentration of intracellular Ca
2+ is low, MICU1−MICU2 inhibits Ca
2+ from entering the mitochondria through the MCU. When cells are stimulated by signals and the concentration of Ca
2+ in the cytoplasm increases and exceeds a certain threshold (more than about 1 mM), MICU1−MICU2 allows Ca
2+ to enter the mitochondria through the MCU [
48]. Down regulation of MICU1 can reduce Ca
2+ flux, decrease mitochondrial oxidative phosphorylation (OXPHOS) and ATP production, and activate AMPK-dependent autophagy [
49]. In parallel, MICU1 also regulates the cristae junction to maintain the structural mitochondrial membrane framework, and without the cristae junction, it can mediate uncoupling and increase ROS production [
50,
51].
MICU1 is upregulated in ovarian cancer cells and its expression is closely related to the survival of cancer cells and tumor growth [
52]. In this pathway, MICU1 induces the accumulation of mitochondrial Ca
2+ and the production of ROS, suggesting that the binding of MICU1 to the MCU is necessary for the function of the MCU complex and the entry of Ca
2+ into mitochondria is a prerequisite survival factor of cancer cells. MICU1 has been shown to be methylated by protein arginine methyltransferase 1 (PRMT1) in cancer cells, yielding decreased Ca
2+ sensitivity and reduced Ca
2+ entry. UCP2/3 is fundamental for mitochondrial Ca
2+ uptake in cancer cells [
53]. When it binds to methylated MICU1, it can normalize the Ca
2+ sensitivity of MICU1 and re-establishes Ca
2+ entry into mitochondria [
54]. This mechanism has also been found to be important in human cancer [
55,
56]. MICU2 can interact with MICU1 and elevate the Ca
2+ threshold activated by the MCU. Therefore, MICU2 can inhibit MCU activity at low Ca
2+ concentrations [
57].
Although MICU1, MICU2, and MICU3 belong to the same family, they have different effects on the MCU. MICU2 is the gatekeeper of the MCU, while MICU3 is an MCU activation enhancer. Overexpression of MICU3 causes a 10-fold increase in transient Ca
2+ [
58]. MCUb directly interacts with the MCU and mainly performs negative regulation of the MCU [
59]. At present, the research results on the effect of the MCUR1 on the MCU are still controversial. Some studies have pointed out that the MCUR1, as an essential scaffold factor of the MCU complex [
60], is the key component of the MCU complex. It has also been reported that mitochondrial Ca
2+ uptake does not depend on the MCUR1, which is only a regulator that sets the Ca
2+ threshold of the transition in mitochondrial permeability. Inhibiting the expression of the MCUR1 increases the Ca
2+ threshold for inducing MPTP conversion, which can reduce the mitochondrial cell death that is induced by an overload of Ca
2+ [
61].
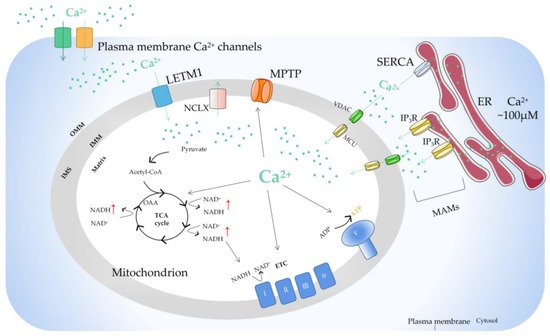
There is a sodium calcium transporter NCLX in the inner membrane of mitochondria, which is a sodium ion (Na
+)-dependent Na
+−Ca
2+ reverse exchange channel and can positively regulate the outflow of Ca
2+ in mitochondria [
62]. When the concentration of Ca
2+ in mitochondria is too high, it will enhance the activity of the NCLX and cause the opening of the MPTP on the inner membrane of mitochondria. Ca
2+ is the center that regulates the MPTP. It can directly regulate the MPTP itself and indirectly affect the MPTP by regulating the adenosine diphosphate (ADP)/ATP balance, mitochondrial membrane potential, and ROS/reactive nitrogen level [
63]. The study found that the MPTP has an important property: the increase in ADP and the recovery of Mg
2+/Ca
2+ caused by MPTP opening are reversible [
64]. This reversibility makes MPTP opening have two modes: continuous opening and instantaneous opening, which can start the cell death signal pathway or maintain the normal physiological function of cells. In addition, there is LETM1 in the mitochondrial inner membrane [
65]. When the concentration of Ca
2+ in the mitochondrial matrix is low, LETM1 can transport Ca
2+ into the matrix. On the contrary, Ca
2+ is transported out of mitochondria. The study also found that silencing LETM1, despite the presence of the MCU, can still inhibit the influx of Ca
2+ into mitochondria (
Figure 2).
Figure 2. The basic mechanism of mitochondrial Ca2+ regulation. Ca2+ transfer from ER to mitochondria occurs on the MAMs, where there are special Ca2+ channels. The opening of IP3R on the surface of ER results in the release of Ca2+ from the lumen of ER. Ca2+ passes through OMM via VDAC and traverses IMM via MCU. Stimulus acts by producing Ca2+ mobilization signals, triggering the increase of intracellular Ca2+ concentration. The function of mitochondrial Ca2+ uptake and release are mainly to regulate the matrix Ca2+ level, thus regulating the activity of mitochondrial dehydrogenase, resulting in increased NADH and ATP production. Ca2+ can also activate mitochondrial ETC complexes. In the steady state, Ca2+ entering mitochondria through MCU must exit through one of the mitochondrial Ca2+ efflux mechanisms. ER, endoplasmic reticulum; MAM, mitochondrial-associated ER membrane; IP3R, inositol triphosphate receptor; MCU, mitochondrial calcium uniporter; VDAC, voltage-dependent anion-selective channel; ETC, electron transport chain; OMM, outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane; LETM1, leucine zipper/EF hand-containing transmembrane-1; MPTP, mitochondrial permeability transition pore; NCLX, mitochondrial Na+/Ca2+ exchanger; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase; OAA, oxaloacetic acid; TCA cycle, tricarboxylic acid cycle.
Mitochondrial Ca
2+ homeostasis is unbalanced in tumors because in tumor cells, the cellular microenvironment is remodeled and leads to further mitochondrial Ca
2+ imbalance, which is an adaptive phenomenon of tumors, and the mitochondrial Ca
2+ imbalance will further promote the development of tumors. Some studies have shown that cancer cells can change mitochondrial Ca
2+ homeostasis mainly through the following methods: (1) Ca
2+ exists in a domain formed between the ER and the mitochondria, which is called the mitochondrial-associated membrane (MAM) and controls mitochondrial Ca
2+ homeostasis [
66] (
Figure 2). Cancer cells can remodel their MAMs to affect mitochondrial Ca
2+ homeostasis and promote cell survival, migration, invasion, metastasis, autophagy, and inhibit apoptosis [
67,
68,
69]. (2) Mechano- and proton-sensing proteins may cause an imbalance in Ca
2+ levels in cancer cells [
70]. (3) In cancer cells, the expression and function of the magnesium (Mg
2+) transporter are abnormal. The imbalance of Mg
2+ homeostasis may destroy Ca
2+ homeostasis [
71]. (4) Cancer cells modify the Ca
2+ signaling network by changing the expression and function of cation channels, pumps, or transporters [
72].
This entry is adapted from 10.3390/ijms23126667