 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abdelhakim BOUYAHYA | -- | 3171 | 2022-04-20 00:09:45 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 3175 | 2022-04-20 04:03:14 | | |
Video Upload Options
Histone modification is an essential mark in maintaining cellular memory and, therefore, loss of this mark can lead to tumor transformation. As these epigenetic changes are reversible, the use of molecules that can restore the functions of the enzymes responsible for the changes is therapeutically necessary. Natural molecules, mainly those isolated from medicinal plants, have demonstrated significant inhibitory properties against enzymes related to histone modifications, particularly histone deacetylases (HDACs). Flavonoids, terpenoids, phenolic acids, and alkaloids exert significant inhibitory effects against HDAC and exhibit promising epi-drug properties. This suggests that epi-drugs against HDAC could prevent and treat various human cancers.
1. Introduction
2. Epigenetic Regulation and Cancer
2.1. Cancer and DNA Methylation
2.2. Oncohistones and Histone Changes
2.2.1. Histone Methylation and Cancer
2.2.2. Histone Acetylation/Deacetylation and Cancer
2.2.3. Phosphorylation, Ubiquitination, SUMOylation, and Cancer
2.2.4. Epigenetic Regulation by miRNAs and Cancer
3. The Role of HDAC in Cancer
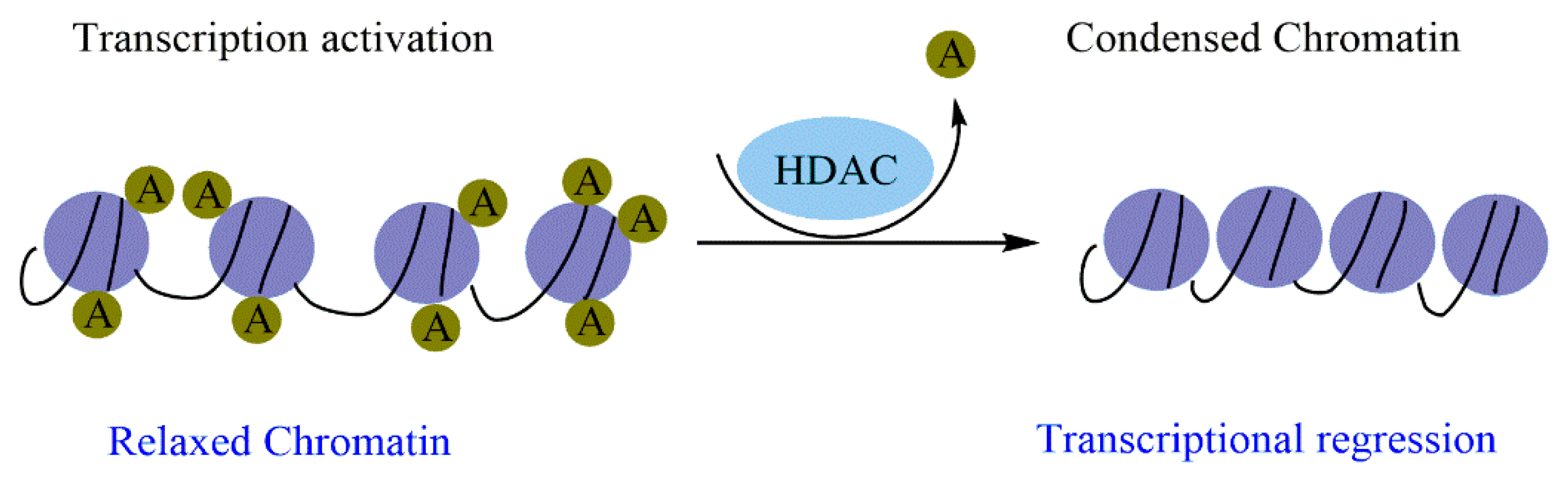
3.1. HDAC in Different Cancer Stages
3.1.1. Cell Cycle Progression and Apoptosis
3.1.2. Differentiation
3.1.3. DNA Damage Response
3.1.4. Metastasis
3.1.5. Angiogenesis
3.1.6. Autophagy
References
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348.
- Bouyahya, A.; El Menyiy, N.; Oumeslakht, L.; El Allam, A.; Balahbib, A.; Rauf, A.; Muhammad, N.; Kuznetsova, E.; Derkho, M.; Thiruvengadam, M.; et al. Preclinical and Clinical Antioxidant Effects of Natural Compounds against Oxidative Stress-Induced Epigenetic Instability in Tumor Cells. Antioxidants 2021, 10, 1553.
- Baylin, S.B.; Jones, P.A. A Decade of Exploring the Cancer Epigenome—Biological and Translational Implications. Nat. Rev. Cancer 2011, 11, 726–734.
- Sandoval, J.; Esteller, M. Cancer Epigenomics: Beyond Genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55.
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692.
- Kanwal, R.; Gupta, S. Epigenetics and Cancer. J. Appl. Physiol. 2010, 109, 598–605.
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20.
- Pfister, S.X.; Ashworth, A. Marked for Death: Targeting Epigenetic Changes in Cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263.
- Holliday, R. DNA Methylation and Epigenetic Inheritance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 326, 329–338.
- Kristensen, L.S.; Nielsen, H.M.; Hansen, L.L. Epigenetics and Cancer Treatment. Eur. J. Pharmacol. 2009, 625, 131–142.
- Karimzadeh, M.R.; Pourdavoud, P.; Ehtesham, N.; Qadbeigi, M.; Asl, M.M.; Alani, B.; Mosallaei, M.; Pakzad, B. Regulation of DNA Methylation Machinery by Epi-MiRNAs in Human Cancer: Emerging New Targets in Cancer Therapy. Cancer Gene Ther. 2021, 28, 157–174.
- Ross, J.P.; Rand, K.N.; Molloy, P.L. Hypomethylation of Repeated DNA Sequences in Cancer. Epigenomics 2010, 2, 245–269.
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal Instability and Tumors Promoted by DNA Hypomethylation. Science 2003, 300, 455.
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A Gene Hypermethylation Profile of Human Cancer. Cancer Res. 2001, 61, 3225–3229.
- He, J.; Zhou, M.; Li, X.; Gu, S.; Cao, Y.; Xing, T.; Chen, W.; Chu, C.; Gu, F.; Zhou, J. SLC34A2 Simultaneously Promotes Papillary Thyroid Carcinoma Growth and Invasion through Distinct Mechanisms. Oncogene 2020, 39, 2658–2675.
- Sastry, N.G.; Wan, X.; Huang, T.; Alvarez, A.A.; Pangeni, R.P.; Song, X.; James, C.D.; Horbinski, C.M.; Brennan, C.W.; Nakano, I. LY6K Promotes Glioblastoma Tumorigenicity via CAV-1–Mediated ERK1/2 Signaling Enhancement. Neuro-Oncol. 2020, 22, 1315–1326.
- Xiao, C.; Wu, G.; Zhou, Z.; Zhang, X.; Wang, Y.; Song, G.; Ding, E.; Sun, X.; Zhong, L.; Li, S. RBBP6, a RING Finger-Domain E3 Ubiquitin Ligase, Induces Epithelial–Mesenchymal Transition and Promotes Metastasis of Colorectal Cancer. Cell Death Dis. 2019, 10, 833.
- Eads, C.A.; Lord, R.V.; Kurumboor, S.K.; Wickramasinghe, K.; Skinner, M.L.; Long, T.I.; Peters, J.H.; DeMeester, T.R.; Danenberg, K.D.; Danenberg, P.V. Fields of Aberrant CpG Island Hypermethylation in Barrett’s Esophagus and Associated Adenocarcinoma. Cancer Res. 2000, 60, 5021–5026.
- Issa, J.-P.J.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated Age-Related CpG Island Methylation in Ulcerative Colitis. Cancer Res. 2001, 61, 3573–3577.
- Greger, V.; Passarge, E.; Höpping, W.; Messmer, E.; Horsthemke, B. Epigenetic Changes May Contribute to the Formation and Spontaneous Regression of Retinoblastoma. Hum. Genet. 1989, 83, 155–158.
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M. Silencing of the VHL Tumor-Suppressor Gene by DNA Methylation in Renal Carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704.
- Gonzalez-Zulueta, M.; Bender, C.M.; Yang, A.S.; Nguyen, T.; Beart, R.W.; Van Tornout, J.M.; Jones, P.A. Methylation of the 5′ CpG Island of the P16/CDKN2 Tumor Suppressor Gene in Normal and Transformed Human Tissues Correlates with Gene Silencing. Cancer Res. 1995, 55, 4531–4535.
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the HMLH1 Promoter Correlates with Lack of Expression of HMLH1 in Sporadic Colon Tumors and Mismatch Repair-Defective Human Tumor Cell Lines. Cancer Res. 1997, 57, 808–811.
- Richter, A.M.; Woods, M.L.; Küster, M.M.; Walesch, S.K.; Braun, T.; Boettger, T.; Dammann, R.H. RASSF10 Is Frequently Epigenetically Inactivated in Kidney Cancer and Its Knockout Promotes Neoplasia in Cancer Prone Mice. Oncogene 2020, 39, 3114–3127.
- Yu, Z.; Feng, J.; Wang, W.; Deng, Z.; Zhang, Y.; Xiao, L.; Wang, Z.; Liu, C.; Liu, Q.; Chen, S. The EGFR-ZNF263 Signaling Axis Silences SIX3 in Glioblastoma Epigenetically. Oncogene 2020, 39, 3163–3178.
- Marini, A.; Mirmohammadsadegh, A.; Nambiar, S.; Gustrau, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Inactivation of Tumor Suppressor Genes in Serum of Patients with Cutaneous Melanoma. J. Investig. Dermatol. 2006, 126, 422–431.
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic Silencing of the PTEN Gene in Melanoma. Cancer Res. 2006, 66, 6546–6552.
- Majumdar, S.; Buckles, E.; Estrada, J.; Koochekpour, S. Aberrant DNA Methylation and Prostate Cancer. Curr. Genom. 2011, 12, 486–505.
- Bell, A.; Bell, D.; Weber, R.S.; El-Naggar, A.K. CpG Island Methylation Profiling in Human Salivary Gland Adenoid Cystic Carcinoma. Cancer 2011, 117, 2898–2909.
- He, S.; Wang, F.; Yang, L.; Guo, C.; Wan, R.; Ke, A.; Xu, L.; Hu, G.; Xu, X.; Shen, J. Expression of DNMT1 and DNMT3a Are Regulated by GLI1 in Human Pancreatic Cancer. PLoS ONE 2011, 6, e27684.
- Baylin, S.B.; Ohm, J.E. Epigenetic Gene Silencing in Cancer–a Mechanism for Early Oncogenic Pathway Addiction? Nat. Rev. Cancer 2006, 6, 107–116.
- Markl, I.D.; Cheng, J.; Liang, G.; Shibata, D.; Laird, P.W.; Jones, P.A. Global and Gene-Specific Epigenetic Patterns in Human Bladder Cancer Genomes Are Relatively Stable in Vivo and in Vitro over Time. Cancer Res. 2001, 61, 5875–5884.
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The Expanding Landscape of ‘Oncohistone’Mutations in Human Cancers. Nature 2019, 567, 473–478.
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L. Mutational Landscape of Uterine and Ovarian Carcinosarcomas Implicates Histone Genes in Epithelial–Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M. Driver Mutations in Histone H3. 3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231.
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.S. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253.
- Koelsche, C.; Schrimpf, D.; Tharun, L.; Roth, E.; Sturm, D.; Jones, D.T.; Renker, E.-K.; Sill, M.; Baude, A.; Sahm, F. Histone 3.3 Hotspot Mutations in Conventional Osteosarcomas: A Comprehensive Clinical and Molecular Characterization of Six H3F3A Mutated Cases. Clin. Sarcoma Res. 2017, 7, 9.
- Lutsik, P.; Baude, A.; Mancarella, D.; Öz, S.; Kühn, A.; Toth, R.; Hey, J.; Toprak, U.H.; Lim, J.; Nguyen, V.H. Globally Altered Epigenetic Landscape and Delayed Osteogenic Differentiation in H3. 3-G34W-Mutant Giant Cell Tumor of Bone. Nat. Commun. 2020, 11, 5414.
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting Epigenetic Modifications in Cancer Therapy: Erasing the Roadmap to Cancer. Nat. Med. 2019, 25, 403–418.
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of Acetylation at Lys16 and Trimethylation at Lys20 of Histone H4 Is a Common Hallmark of Human Cancer. Nat. Genet. 2005, 37, 391–400.
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266.
- Prachayasittikul, V.; Prathipati, P.; Pratiwi, R.; Phanus-Umporn, C.; Malik, A.A.; Schaduangrat, N.; Seenprachawong, K.; Wongchitrat, P.; Supokawej, A.; Prachayasittikul, V. Exploring the Epigenetic Drug Discovery Landscape. Expert Opin. Drug Discov. 2017, 12, 345–362.
- Kubicek, S.; Gilbert, J.C.; Fomina-Yadlin, D.; Gitlin, A.D.; Yuan, Y.; Wagner, F.F.; Holson, E.B.; Luo, T.; Lewis, T.A.; Taylor, B. Chromatin-Targeting Small Molecules Cause Class-Specific Transcriptional Changes in Pancreatic Endocrine Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 5364–5369.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837.
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone Deacetylases in Acute Myeloid Leukaemia Show a Distinctive Pattern of Expression That Changes Selectively in Response to Deacetylase Inhibitors. Leukemia 2005, 19, 1751–1759.
- Fang, D.; Gan, H.; Cheng, L.; Lee, J.-H.; Zhou, H.; Sarkaria, J.N.; Daniels, D.J.; Zhang, Z. H3. 3K27M Mutant Proteins Reprogram Epigenome by Sequestering the PRC2 Complex to Poised Enhancers. Elife 2018, 7, e36696.
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C. Histone H3. 3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.
- Chase, A.; Cross, N.C. Aberrations of EZH2 in Cancer. Clin. Cancer Res. 2011, 17, 2613–2618.
- Fang, D.; Gan, H.; Lee, J.-H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J. The Histone H3. 3K36M Mutation Reprograms the Epigenome of Chondroblastomas. Science 2016, 352, 1344–1348.
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R. Histone H3K36 Mutations Promote Sarcomagenesis through Altered Histone Methylation Landscape. Science 2016, 352, 844–849.
- Lhoumaud, P.; Badri, S.; Rodriguez-Hernaez, J.; Sakellaropoulos, T.; Sethia, G.; Kloetgen, A.; Cornwell, M.; Bhattacharyya, S.; Ay, F.; Bonneau, R. NSD2 Overexpression Drives Clustered Chromatin and Transcriptional Changes in a Subset of Insulated Domains. Nat. Commun. 2019, 10, 4843.
- Karatas, H.; Townsend, E.C.; Cao, F.; Chen, Y.; Bernard, D.; Liu, L.; Lei, M.; Dou, Y.; Wang, S. High-Affinity, Small-Molecule Peptidomimetic Inhibitors of MLL1/WDR5 Protein–Protein Interaction. J. Am. Chem. Soc. 2013, 135, 669–682.
- Xhemalce, B.; Dawson, M.A.; Bannister, A.J. Histone Modifications. In Reviews in Cell Biology and Molecular Medicine; Wiley: Hoboken, NJ, USA, 2006.
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and Mechanisms of Non-Histone Protein Acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174.
- Marmorstein, R. Structure of Histone Acetyltransferases. J. Mol. Biol. 2001, 311, 433–444.
- Calcagno, D.Q.; Wisnieski, F.; Mota, E.R.; Maia de Sousa, S.B.; Costa da Silva, J.M.; Leal, M.F.; Gigek, C.O.; Santos, L.C.; Rasmussen, L.T.; Assumpção, P.P. Role of Histone Acetylation in Gastric Cancer: Implications of Dietetic Compounds and Clinical Perspectives. Epigenomics 2019, 11, 349–362.
- Gruber, J.J.; Geller, B.; Lipchik, A.M.; Chen, J.; Salahudeen, A.A.; Ram, A.N.; Ford, J.M.; Kuo, C.J.; Snyder, M.P. HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Mol. Cell 2019, 75, 711–724.
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Bian, J.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Correction: Role of Novel Histone Modifications in Cancer. Oncotarget 2018, 9, 19460.
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J. Inactivating Mutations of Acetyltransferase Genes in B-Cell Lymphoma. Nature 2011, 471, 189–195.
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 Pathway Facilitates Nuclear Accumulation of HIF-1α and Functional Activation of HIF-1 by Deacetylating Hsp70 in the Cytosol. Cell Cycle 2015, 14, 2520–2536.
- Li, J.; Huang, C.; Xiong, T.; Zhuang, C.; Zhuang, C.; Li, Y.; Ye, J.; Gui, Y. A CRISPR Interference of CBP and P300 Selectively Induced Synthetic Lethality in Bladder Cancer Cells In Vitro. Int. J. Biol. Sci. 2019, 15, 1276.
- Davis, P.K.; Brachmann, R.K. Chromatin Remodeling and Cancer. Cancer Biol. Ther. 2003, 2, 23–30.
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840.
- Cohen, P. The Origins of Protein Phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130.
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten Things You Should Know about Protein Kinases: IUPHAR R Eview 14. Br. J. Pharmacol. 2015, 172, 2675–2700.
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428.
- Seeler, J.-S.; Dejean, A. SUMO and the Robustness of Cancer. Nat. Rev. Cancer 2017, 17, 184–197.
- Ryan, B.M.; Robles, A.I.; Harris, C.C. Genetic Variation in MicroRNA Networks: The Implications for Cancer Research. Nat. Rev. Cancer 2010, 10, 389–402.
- Kasinski, A.L.; Slack, F.J. MiRNA-34 Prevents Cancer Initiation and Progression in a Therapeutically Resistant K-Ras and P53-Induced Mouse Model of Lung Adenocarcinoma. Cancer Res. 2012, 72, 5576–5587.
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269.
- Lin, H.-Y.; Chen, C.-S.; Lin, S.-P.; Weng, J.-R.; Chen, C.-S. Targeting Histone Deacetylase in Cancer Therapy. Med. Res. Rev. 2006, 26, 397–413.
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of Histone Deacetylases in Epigenetic Regulation: Emerging Paradigms from Studies with Inhibitors. Clin. Epigenetics 2012, 4, 5.
- Ropero, S.; Esteller, M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25.
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone Deacetylases and Epigenetic Therapies of Hematological Malignancies. Pharmacol. Res. 2010, 62, 18–34.
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. Prostate 2004, 59, 177–189.
- Wilson, A.J.; Byun, D.-S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone Deacetylase 3 (HDAC3) and Other Class I HDACs Regulate Colon Cell Maturation and P21 Expression and Are Deregulated in Human Colon Cancer. J. Biol. Chem. 2006, 281, 13548–13558.
- Choi, J.-H.; Kwon, H.J.; Yoon, B.-I.; Kim, J.-H.; Han, S.U.; Joo, H.J.; Kim, D.-Y. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304.
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 MRNA Expression in Invasive Carcinoma of the Breast. Breast Cancer Res. Treat. 2005, 94, 11–16.
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.-P.; Göttlicher, M. Induction of HDAC2 Expression upon Loss of APC in Colorectal Tumorigenesis. Cancer Cell 2004, 5, 455–463.
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of Histone Deacetylase 2 Increases Apoptosis and P21 Cip1/WAF1 Expression, Independent of Histone Deacetylase 1. Cell Death Differ. 2005, 12, 395–404.
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y. Increased Expression of Histone Deacetylase 2 Is Found in Human Gastric Cancer. Apmis 2005, 113, 264–268.
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968.
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F. A Truncating Mutation of HDAC2 in Human Cancers Confers Resistance to Histone Deacetylase Inhibition. Nat. Genet. 2006, 38, 566–569.
- Zimmermann, S.; Kiefer, F.; Prudenziati, M.; Spiller, C.; Hansen, J.; Floss, T.; Wurst, W.; Minucci, S.; Göttlicher, M. Reduced Body Size and Decreased Intestinal Tumor Rates in HDAC2-Mutant Mice. Cancer Res. 2007, 67, 9047–9054.
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.-W.; Hiebert, S.W. Deletion of Histone Deacetylase 3 Reveals Critical Roles in S Phase Progression and DNA Damage Control. Mol. Cell 2008, 30, 61–72.
- Glaser, K.B.; Li, J.; Staver, M.J.; Wei, R.-Q.; Albert, D.H.; Davidsen, S.K. Role of Class I and Class II Histone Deacetylases in Carcinoma Cells Using SiRNA. Biochem. Biophys. Res. Commun. 2003, 310, 529–536.
- Inoue, S.; Mai, A.; Dyer, M.J.; Cohen, G.M. Inhibition of Histone Deacetylase Class I but Not Class II Is Critical for the Sensitization of Leukemic Cells to Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand–Induced Apoptosis. Cancer Res. 2006, 66, 6785–6792.
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.-C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I Histone Deacetylase Expression Has Independent Prognostic Impact in Human Colorectal Cancer: Specific Role of Class I Histone Deacetylases in Vitro and in Vivo. Clin. Cancer Res. 2008, 14, 1669–1677.
- Wilson, A.J.; Byun, D.-S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H. HDAC4 Promotes Growth of Colon Cancer Cells via Repression of P21. Mol. Biol. Cell 2008, 19, 4062–4075.
- Park, J.-H.; Kim, S.-H.; Choi, M.-C.; Lee, J.; Oh, D.-Y.; Im, S.-A.; Bang, Y.-J.; Kim, T.-Y. Class II Histone Deacetylases Play Pivotal Roles in Heat Shock Protein 90-Mediated Proteasomal Degradation of Vascular Endothelial Growth Factor Receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322.
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-ΚB-Dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 2004, 23, 2369–2380.
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P. Composition and Histone Substrates of Polycomb Repressive Group Complexes Change during Cellular Differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864.
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O. HDAC6 Rescues Neurodegeneration and Provides an Essential Link between Autophagy and the UPS. Nature 2007, 447, 860–864.
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799.
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The Mammalian SIR2α Protein Has a Role in Embryogenesis and Gametogenesis. Mol. Cell. Biol. 2003, 23, 38–54.
- Wang, R.-H.; Sengupta, K.; Li, C.; Kim, H.-S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell 2008, 14, 312–323.
- Saunders, L.R.; Verdin, E. Sirtuins: Critical Regulators at the Crossroads between Cancer and Aging. Oncogene 2007, 26, 5489–5504.
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A. Proteomics-Based Identification of Differentially Expressed Genes in Human Gliomas: Down-Regulation of SIRT2 Gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566.
- Michan, S.; Sinclair, D. Sirtuins in Mammals: Insights into Their Biological Function. Biochem. J. 2007, 404, 1–13.
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The Cytoplasmic Deacetylase HDAC6 Is Required for Efficient Oncogenic Tumorigenesis. Cancer Res. 2008, 68, 7561–7569.
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcene, A.; Verdin, E.; Castronovo, V. HDAC4 Represses P21 WAF1/Cip1 Expression in Human Cancer Cells through a Sp1-Dependent, P53-Independent Mechanism. Oncogene 2009, 28, 243–256.
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C. Role for Histone Deacetylase 1 in Human Tumor Cell Proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795.
- Draney, C.; Austin, M.C.; Leifer, A.H.; Smith, C.J.; Kener, K.B.; Aitken, T.J.; Hess, K.H.; Haines, A.C.; Lett, L.A.; Hernandez-Carretero, A. HDAC1 Overexpression Enhances β-Cell Proliferation by down-Regulating Cdkn1b/P27. Biochem. J. 2018, 475, 3997–4010.
- Zhao, H.; Wang, Y.; Yang, C.; Zhou, J.; Wang, L.; Yi, K.; Li, Y.; Shi, J.; Kang, C.; Zeng, L. EGFR-VIII Downregulated H2AZK4/7AC Though the PI3K/AKT-HDAC2 Axis to Regulate Cell Cycle Progression. Clin. Transl. Med. 2020, 9, 10.
- Wei, D.; Lu, T.; Ma, D.; Yu, K.; Zhang, T.; Xiong, J.; Wang, W.; Zhang, Z.; Fang, Q.; Wang, J. Synergistic Activity of Imatinib and AR-42 against Chronic Myeloid Leukemia Cells Mainly through HDAC1 Inhibition. Life Sci. 2018, 211, 224–237.
- Jiang, Y.; Hsieh, J. HDAC3 Controls Gap 2/Mitosis Progression in Adult Neural Stem/Progenitor Cells by Regulating CDK1 Levels. Proc. Natl. Acad. Sci. USA 2014, 111, 13541–13546.
- Liu, D.-L.; Lu, L.-L.; Dong, L.-L.; Liu, Y.; Bian, X.-Y.; Lian, B.-F.; Xie, L.; Wen, D.; Gao, D.-M.; Ke, A.-W. MiR-17-5p and MiR-20a-5p Suppress Postoperative Metastasis of Hepatocellular Carcinoma via Blocking HGF/ERBB3-NF-ΚB Positive Feedback Loop. Theranostics 2020, 10, 3668–3683.
- Yang, J.; Li, B.; Zhao, S.; Du, H.; Du, Y. Exosomal MiR-638 Inhibits Hepatocellular Carcinoma Progression by Targeting SP1. OncoTargets Ther. 2020, 13, 6709–6720.
- Chen, M.-K.; Cai, M.-Y.; Luo, R.-Z.; Tian, X.; Liao, Q.-M.; Zhang, X.-Y.; Han, J.-D. Overexpression of P300 Correlates with Poor Prognosis in Patients with Cutaneous Squamous Cell Carcinoma. Br. J. Dermatol. 2015, 172, 111–119.
- Lin, J.; Liu, Z.; Liao, S.; Li, E.; Wu, X.; Zeng, W. Elevated MicroRNA-7 Inhibits Proliferation and Tumor Angiogenesis and Promotes Apoptosis of Gastric Cancer Cells via Repression of Raf-1. Cell Cycle 2020, 19, 2496–2508.
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.-H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A. Histone Deacetylase 11 Inhibition Promotes Breast Cancer Metastasis from Lymph Nodes. Nat. Commun. 2019, 10, 4192.
- Patra, S.; Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Jena, M.; Bhutia, S.K. Dysregulation of Histone Deacetylases in Carcinogenesis and Tumor Progression: A Possible Link to Apoptosis and Autophagy. Cell. Mol. Life Sci. 2019, 76, 3263–3282.
- Atsumi, A.; Tomita, A.; Kiyoi, H.; Naoe, T. Histone Deacetylase 3 (HDAC3) Is Recruited to Target Promoters by PML-RARα as a Component of the N-CoR Co-Repressor Complex to Repress Transcription in Vivo. Biochem. Biophys. Res. Commun. 2006, 345, 1471–1480.
- Chauchereau, A.; Mathieu, M.; de Saintignon, J.; Ferreira, R.; Pritchard, L.L.; Mishal, Z.; Dejean, A.; Harel-Bellan, A. HDAC4 Mediates Transcriptional Repression by the Acute Promyelocytic Leukaemia-Associated Protein PLZF. Oncogene 2004, 23, 8777–8784.
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, Fusion Partner in t (8; 21) Acute Myeloid Leukemia, Represses Transcription by Interaction with the Human N-CoR/MSin3/HDAC1 Complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865.
- Amann, J.M.; Nip, J.; Strom, D.K.; Lutterbach, B.; Harada, H.; Lenny, N.; Downing, J.R.; Meyers, S.; Hiebert, S.W. ETO, a Target of t (8; 21) in Acute Leukemia, Makes Distinct Contacts with Multiple Histone Deacetylases and Binds MSin3A through Its Oligomerization Domain. Mol. Cell. Biol. 2001, 21, 6470–6483.
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99.
- Kolbinger, F.R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A.K. The HDAC6/8/10 Inhibitor TH34 Induces DNA Damage-Mediated Cell Death in Human High-Grade Neuroblastoma Cell Lines. Arch. Toxicol. 2018, 92, 2649–2664.
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151.
- Nishimoto, K.; Niida, H.; Uchida, C.; Ohhata, T.; Kitagawa, K.; Motegi, A.; Suda, T.; Kitagawa, M. HDAC3 Is Required for XPC Recruitment and Nucleotide Excision Repair of DNA Damage Induced by UV Irradiation. Mol. Cancer Res. 2020, 18, 1367–1378.
- Liu, X.; Wang, Y.; Zhang, R.; Jin, T.; Qu, L.; Jin, Q.; Zheng, J.; Sun, J.; Wu, Z.; Wang, L. HDAC10 Is Positively Associated with PD-L1 Expression and Poor Prognosis in Patients with NSCLC. Front. Oncol. 2020, 10, 485.
- Yang, W.-C.; Bao, H.-Y.; Liu, Y.-Y.; Nie, Y.-Y.; Yang, J.-M.; Hong, P.-Z.; Zhang, Y. Depsidone Derivatives and a Cyclopeptide Produced by Marine Fungus Aspergillus Unguis under Chemical Induction and by Its Plasma Induced Mutant. Molecules 2018, 23, 2245.
- Zhang, M.; Xiang, S.; Joo, H.-Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C. HDAC6 Deacetylates and Ubiquitinates MSH2 to Maintain Proper Levels of MutSα. Mol. Cell 2014, 55, 31–46.
- Lagunas-Rangel, F.A. Current Role of Mammalian Sirtuins in DNA Repair. DNA Repair 2019, 80, 85–92.
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of P53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148.
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 Promotes DNA Repair under Stress by Activating PARP1. Science 2011, 332, 1443–1446.
- Van Meter, M.; Simon, M.; Tombline, G.; May, A.; Morello, T.D.; Hubbard, B.P.; Bredbenner, K.; Park, R.; Sinclair, D.A.; Bohr, V.A. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep. 2016, 16, 2641–2650.
- Zhang, H.; Chen, S.; Jia, X.; Huang, Y.; Ji, R.; Zhao, L. Comparation of the Phytotoxicity between Chemically and Green Synthesized Silver Nanoparticles. Sci. Total Environ. 2021, 752, 142264.
- Moustakas, A.; Heldin, C.-H. Signaling Networks Guiding Epithelial–Mesenchymal Transitions during Embryogenesis and Cancer Progression. Cancer Sci. 2007, 98, 1512–1520.
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.-H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235.
- Li, W.; Ding, X.; Wang, S.; Xu, L.; Yin, T.; Han, S.; Geng, J.; Sun, W. Downregulation of Serum Exosomal MiR-320d Predicts Poor Prognosis in Hepatocellular Carcinoma. J. Clin. Lab. Anal. 2020, 34, e23239.
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 Induces EMT by Cooperating with EMT Transcription Factors and Enhances Prostate Cancer Cell Migration and Metastasis. Oncogene 2012, 31, 4619–4629.
- Cheng, X.; Hung, M.-C. Breast Cancer Brain Metastases. Cancer Metastasis Rev. 2007, 26, 635–643.
- Carmeliet, P. Angiogenesis in Life, Disease and Medicine. Nature 2005, 438, 932–936.
- Moehler, T.; Neben, K.; Ho, A.; Goldschmidt, H. Angiogenesis in Hematologic Malignancies. Ann. Hematol. 2001, 80, 695–705.
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.-E.; Lee, S.-W.; Moon, E.-J.; Kim, H.-S.; Lee, S.-K.; Chung, H.Y. Histone Deacetylases Induce Angiogenesis by Negative Regulation of Tumor Suppressor Genes. Nat. Med. 2001, 7, 437–443.
- Hrgovic, I.; Doll, M.; Pinter, A.; Kaufmann, R.; Kippenberger, S.; Meissner, M. Histone Deacetylase Inhibitors Interfere with Angiogenesis by Decreasing Endothelial VEGFR-2 Protein Half-Life in Part via a VE-Cadherin-Dependent Mechanism. Exp. Dermatol. 2017, 26, 194–201.
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs Mediate HIF-1α Stability Through PHD2-Dependent Mechanism, While HDAC6, a Class IIb Member, Promotes HIF-1α Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. J. Bone Miner. Res. 2016, 31, 1287–1299.
- Urbich, C.; Rössig, L.; Kaluza, D.; Potente, M.; Boeckel, J.-N.; Knau, A.; Diehl, F.; Geng, J.-G.; Hofmann, W.-K.; Zeiher, A.M. HDAC5 Is a Repressor of Angiogenesis and Determines the Angiogenic Gene Expression Pattern of Endothelial Cells. Blood J. Am. Soc. Hematol. 2009, 113, 5669–5679.
- Tsou, P.-S.; Khanna, D.; Sawalha, A.H. Identification of Cysteine-Rich Angiogenic Inducer 61 as a Potential Antifibrotic and Proangiogenic Mediator in Scleroderma. Arthritis Rheumatol. 2019, 71, 1350–1359.
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.-P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.G. Class IIb HDAC6 Regulates Endothelial Cell Migration and Angiogenesis by Deacetylation of Cortactin. EMBO J. 2011, 30, 4142–4156.
- Cecconi, F.; Levine, B. The Role of Autophagy in Mammalian Development: Cell Makeover Rather than Cell Death. Dev. Cell 2008, 15, 344–357.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylases 1 and 2 Regulate Autophagy Flux and Skeletal Muscle Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654.
- Majora, M.; Sondenheimer, K.; Knechten, M.; Uthe, I.; Esser, C.; Schiavi, A.; Ventura, N.; Krutmann, J. HDAC Inhibition Improves Autophagic and Lysosomal Function to Prevent Loss of Subcutaneous Fat in a Mouse Model of Cockayne Syndrome. Sci. Transl. Med. 2018, 10, eaam7510.
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218.
- Lee, J.-Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.-P. Disease-Causing Mutations in Parkin Impair Mitochondrial Ubiquitination, Aggregation, and HDAC6-Dependent Mitophagy. J. Cell Biol. 2010, 189, 671–679.
- Lee, K.J.; Lee, K.Y.; Lee, Y.M. Downregulation of a Tumor Suppressor RECK by Hypoxia through Recruitment of HDAC1 and HIF-1α to Reverse HRE Site in the Promoter. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2010, 1803, 608–616.
- Oehme, I.; Linke, J.-P.; Böck, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M. Histone Deacetylase 10 Promotes Autophagy-Mediated Cell Survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601.
- Garva, R.; Thepmalee, C.; Yasamut, U.; Sudsaward, S.; Guazzelli, A.; Rajendran, R.; Tongmuang, N.; Khunchai, S.; Meysami, P.; Limjindaporn, T. Sirtuin Family Members Selectively Regulate Autophagy in Osteosarcoma and Mesothelioma Cells in Response to Cellular Stress. Front. Oncol. 2019, 9, 949.
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466.
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 Positively Regulates Autophagy and Mitochondria Function in Embryonic Stem Cells under Oxidative Stress. Stem Cells 2014, 32, 1183–1194.
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L. SIRT5 Regulation of Ammonia-Induced Autophagy and Mitophagy. Autophagy 2015, 11, 253–270.
- Shi, L.; Yan, H.; An, S.; Shen, M.; Jia, W.; Zhang, R.; Zhao, L.; Huang, G.; Liu, J. SIRT 5-Mediated Deacetylation of LDHB Promotes Autophagy and Tumorigenesis in Colorectal Cancer. Mol. Oncol. 2019, 13, 358–375.
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury through Targeting Nkx3. 2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461.

