Histone modification is an essential mark in maintaining cellular memory and, therefore, loss of this mark can lead to tumor transformation. As these epigenetic changes are reversible, the use of molecules that can restore the functions of the enzymes responsible for the changes is therapeutically necessary. Natural molecules, mainly those isolated from medicinal plants, have demonstrated significant inhibitory properties against enzymes related to histone modifications, particularly histone deacetylases (HDACs). Flavonoids, terpenoids, phenolic acids, and alkaloids exert significant inhibitory effects against HDAC and exhibit promising epi-drug properties. This suggests that epi-drugs against HDAC could prevent and treat various human cancers.
1. Introduction
Cancer is a group of pathologies with severe consequences for human health. It is induced by numerous risk factors, such as genetic predisposition, hormonal disorders, oxidative stress, epigenetic instability, microbial infections, and many others [
1]. Recently, close correlations between loss of cellular memory, epigenetic instability, and the onset of certain cancers have been demonstrated [
2]. In this respect, epigenetic modifications are biochemical marks deposited on DNA and histones, and they participate in the regulation of gene expression at the transcriptional level. These modifications have several roles, including maintaining cell memory during mitotic division; differentiated cells must retain their cellular memory to keep their identity. However, cells can lose their memory and become cancerous due to epigenetic instability. HDACs play a key role in maintaining repressive gene activity. The repressive activity of HDAC leads to the ectopic repression of various genes, which can affect cellular memory. In addition, normal cells can become cancerous after transformation.
2. Epigenetic Regulation and Cancer
The involvement of environmental, lifestyle, and hereditary factors is the origin of the complexity of cancer etiology. Cancer is most commonly considered a genetic pathology induced by gene expression alteration, following repetitive genetic aberrations. Currently, it is known that the disruption of gene expression causing cell transformation is controlled by epigenetics. Thus, the abnormality of epigenetic regulation has become a model to explain carcinogenesis and cancer development. Research findings suggest that genetic and epigenetic mechanisms are not separate events in cancer, but interact and benefit from each other during tumorigenesis [
17,
18]. Epigenetic changes are labeled as ‘first hits’ for tumorigeneses. They are the early events responsible for the loss of tissular homeostasis and induce genetic instability; thus, causing changes in the expression profile of tumor-suppressor genes. Furthermore, it is evident that several tumor suppressor genes are rarely genetically mutated but epigenetically silenced [
19].
Epigenetic events include histone modifications (phosphorylation, acetylation, methylation, SUMOylation, and ubiquitylation), DNA methylation, and deregulation of non-coding RNAs and their interactions with proteins or nucleic acids [
20]. The dynamic regulation of histone marks, DNA, and chromatin structure is dynamically performed by four types of epigenetic regulators, including (i) writers, (ii) erasers, (iii) readers, and (iv) remodelers [
21]. Deregulation and mutations in the genes encoding these epigenetic regulators have been described in various cancers [
22].
2.1. Cancer and DNA Methylation
DNA methylation consisting of a covalent transfer is the most commonly studied epigenetic modification by DNA methyltransferases (DNMTs) of methyl groups at the fifth carbon of cytosine (5-mC) within 5′-CpG-3′ di-nucleotides [
23,
24]. In mammals, DNMT1, DNMT3a, and DNMT3b are the three main classes of DNMT enzymes. In this regard, abnormal DNA methylation patterns are due to DNMT overexpression or aberrant recruitment. Cancer cells exhibit abnormal DNA methylation patterns, marked by global hypomethylation associated with the promoter and focal hypermethylation of specific genes [
25,
26]. In addition, aberrant hypomethylation induces the expression of numerous genes, including oncogenes [
27], whereas hypermethylation inhibits specific tumor suppressor genes (TSGs) [
28]. On the other hand, hypomethylation of oncogenesis is often revealed in cancers such as SLC34A2 in papillary thyroid carcinoma, LY6K in glioblastoma, and RBBP6 in colorectal cancer, among others [
29,
30,
31].
Moreover, hypermethylation is easily observed at precancerous stages in benign tumors and tumor-predisposing inflammatory lesions [
32,
33]. In this respect, the retinoblastoma gene’s hypermethylation of the CpG island promoter is released in retinoblastoma [
34]. Several research studies have shown promoter hypermethylation and silencing of other TSGs in renal cancer;
VHL (
von Hippel–Lindau) [
35] in bladder cancer; the cell cycle regulator
CDKN2 A/p16 [
36]; and in colon cancer, the mismatch repair gene
hMLH1 [
37]. Additionally, abundant hypermethylated TSGs include RASSF10 and SIX3 in kidney cancer and glioblastoma, respectively [
38,
39]; PTEN, and CDKN2A in melanoma [
40,
41]; and CDKN2A, TIMPS, and DAPK in prostate cancer [
42]. In contrast, significant methylation of EN1 and SCTR was observed in the prostate, colorectal, and salivary gland adenoid cystic carcinoma [
43]. The gene APC is hypermethylated in pancreatic cancer [
44]. Indeed, hMLH1 still carries a genetic mutation and hypermethylation of an allele in the colon cancer cell line HCT116, which induces the inactivation of the main tumor suppressors [
45]. Moreover, pax6, p16, and p15 are generally aberrantly methylated in bladder cancer [
46].
2.2. Oncohistones and Histone Changes
Histone mutations play an important role in cancer epigenetics, where recurrent mutations targeting histone genes have been described in several types of cancer. Genes coding for histones are mostly mutated in cancer and are commonly named ‘oncohistones’ [
47]. Mutations affecting all canonical histone classes and non-canonical histones have been recorded in different tumors [
47]. H2A and H2B mutations occur in carcinosarcomas, while H1 mutations were detected in diffuse large B cell lymphomas [
47,
48]. Similarly, mutations in H3 and its non-canonical counterpart H3.3 were seen in children’s tumors [
49,
50], whereas mutations in osteosarcoma and giant cell tumor of bone harbor H3.3 G34W mutations [
51,
52].
Histone modifications control chromatin’s active and inactive states, which ultimately affect gene expression [
53]. Important roles are attributed to certain histone modifications in epigenetic acetylation, methylation, and deregulation; linked to epigenetic abnormalities in cancer cells [
54,
55]. In core histones, H3 and H4, methylation of specific residues and loss of acetylation have been defined as a marker of cancer cells [
54,
55]. Enzymes that modulate histone modification include histone demethylase (HDMT), histone methyltransferase (HMT), E3-ubiquitin and kinases, histone acetyltransferase (HAT), and histone deacetylase (HDAC) [
56].
2.2.1. Histone Methylation and Cancer
Histone methylation affects the affinity of transcription factors, leading to activation or restriction of transcription. Irregularities in the methylation of different lysine residues can modify gene expression [
57]. In general, H3K4, H3K36, and H3K79 are the most important sites where their methylation causes gene transcription, while methylation of H3K27, H4K20, and H3K9 is related to transcription silencing [
58]. In addition, trimethylations of H3K79, H3K27, and H3K9 induce repression, while mono-methylations of H4K20, H3K27, H3K9, H2BK5, and H3K79 induce gene expression.
H3K27M has been detected in 20% of pediatric glioblastomas and up to 70% of diffuse intrinsic pontine gliomas (DIPGs), resulting in decreased trimethylation of H3K27 (H3K27me3) [
50,
59,
60,
61]. Degradation of the EZH2-induced methylation of H3K27 has been reported in breast, prostate, bladder, lung, and kidney cancers and hematological malignancies [
62]. Meanwhile, the H3K36M mutation induces undifferentiated sarcoma, causing increased levels of H3K27me3 and global loss of H3K36 (me2 and me) [
63,
64]. In this regard, H3K27ac enrichment and interactions with other regulatory constituents are promoted by H3K36me2 expansion favors; thus, activating oncogenic pathways [
65]. Moreover, increased H3K4me3 is observed in myeloid and lymphoid leukemias [
66].
2.2.2. Histone Acetylation/Deacetylation and Cancer
Histone acetylation and deacetylation are correlated with the active and open chromatin conformation and inactive and condensed chromatin form, respectively. In this respect, the HDAC and HAT enzymes control the regulation of histone acetylation, which is very dynamic [
67]. The acetyl-CoA acetyl group was added, by HAT, to the histone at the lysine position, which neutralizes the positive lysine charge disrupting the electrostatic interaction between DNA and histones. The chromatin structure was attenuated; thus, affecting the gene assembly and changing the transcription operation [
68].
An imbalance of histone acetylation was detected in lung cancers, Rubinstein–Taybi syndrome, AML, and glioblastomas [
69]. Many members of HATs mutate differently in tumors [
70,
71,
72] and participate in different stages of its development, including B-cell non-Hodgkin lymphoma and leukemia [
73], and have also been described in solid cancers [
74], [
75]. Furthermore, chromosomal translocations implying HATs and their fusion proteins were involved in the onset and development of acute leukemia [
76]. Moreover, any disturbance in the expression of distinct HDAC isoforms induces different cancers. Indeed, dysregulation of HDAC proteins causes aberrant deacetylation and inhibition of TSGs. HDACs can also regulate gene transcription via deacetylating the DNMT, HAT, and HDAC proteins responsible for epigenetic events [
77].
2.2.3. Phosphorylation, Ubiquitination, SUMOylation, and Cancer
Abnormalities in kinase activity lead to a variety of cancers [
78,
79]. The decreased activity of E3 ubiquitin ligase caused by specific mutations can be the origin of various tumors, such as breast cancer and renal cell carcinoma. Moreover, cervical cancer is caused by an increase in the ubiquitination effect, while glioblastoma and colorectal cancer are induced by the total elimination of ubiquitination [
80]. SUMOylation is a process required for all cells, and not as a tumor promoter and suppressor [
81].
2.2.4. Epigenetic Regulation by miRNAs and Cancer
Epigenetic linked noncoding RNAs (ncRNAs) comprise small interfering RNA (siRNAs), long noncoding RNAs (lncRNAs), microRNAs (miRNAs), and Piwi-interacting RNA (piRNAs). In this regard, ncRNAs are involved in complex double-negative feedback loops, where miRNA inhibition of an epigenetic regulator is performed at the epigenetic level by the same regulator. In contrast, the epigenetic modifier enzymes involved in epigenetic modulation can be targeted by miRNAs, establishing a trilateral regulatory ‘epi–miR–epi’ feedback circuit. The intricate interaction between the epigenetic architecture and miRNAs is important to surveilling gene expression profiles in cancer [
82]. They have been classified as tumor suppressors, oncogenic, or context-dependent miRNAs [
83]. Furthermore, miRNAs regulate the expression of epigenetic regulatory enzymes, such as HMT, DNMT, and HAT. Moreover, aberrant miRNA profiling indicative of altered regulatory factors, including cell migration and proliferation, has been reported in numerous tumors, with the most manifest being the decreased ncRNA expression levels in cancer cells compared to normal tissues [
84].
3. The Role of HDAC in Cancer
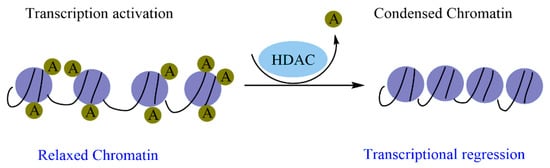
In many human cancers, dysregulation of epigenetic enzymes results from mutations, abnormal expression, and/or disproportionate recruitment to certain loci. Histone acetylation modulates the chromatin structure, which is a key factor regulating gene expression. Histone acetylation is due to the well-balanced activities of HATs and HDACs. Indeed, HDACs remove the acetyl moiety from lysine residues, resulting in positively charged histones. This increases the ionic interactions with DNA that provoke a compacted chromatin structure, which represses gene expression by making it difficult to access the transcription machinery. Moreover, in the absence of a signal, HDACs can form a corepressor complex with the nuclear receptor and interact directly with transcription factors (
Figure 1) [
98].
Figure 1. The catalytic action of HDAC enzymes on chromatin condensation.
So far, 18 mammalian HDACs have been described and assembled into four families according to their homology with yeast HDACs [
99,
100]. Homologous to yeast Rpd3, Class I HDACs (1, 2, 3, and 8) are localized in the nucleus of human cells. Class II HDACs (4, 5, 6, 7, 9, and 10) are homologous to yeast Hda1, exhibit a tissue-specific expression, and can shift between the nucleus and cytoplasm. Class III HDACs, or sirtuins (SIRT1-7), are homologous to yeast Sir2 and require the NAD+ cofactor for their activity. Finally, Class IV HDACs, including the recently discovered HDAC11, display the characteristics of both Class I HDACs and HDAC II [
99,
100].
The implications of HDACs in cancer development were first reported in hematological malignancies by inappropriate involvement of HDAC-containing complexes [
100,
101]. Until now, very rare mutations altering HDAC expression and activity have been recorded in tumors, while deregulation of their activity has been associated with abnormal gene expression and carcinogenesis. Many studies reported that HDAC1 is overexpressed in prostate, colon adenocarcinoma gastric, and breast carcinomas [
102,
103,
104,
105], whereas HDAC2 is overexpressed in colorectal [
106], cervical [
107], and gastric cancers [
108]. Overexpression of HDAC1, HDAC2, and HDAC3 is linked to low survival in patients with gastric and ovarian cancers, while HDAC6 was highly expressed in breast cancer specimens [
109]. HDAC8 overexpression was reported in neuroblastoma, whereas low HDAC4 levels are reported in gastric cancers [
110,
111].
Research findings indicated that knockdown of HDAC genes induced apoptosis and cell cycle arrest, particularly HDAC 1, 2, 3, and 6, in various cancers (colon, breast, lung, and acute promyelocytic leukemia (APL)) [
103,
111,
112,
113,
114,
115]. Knockdown of the HDAC4 gene inhibited cell proliferation and induced apoptosis [
116]. Class II HDACs are also involved in angiogenesis regulation, as the knockdown of HDAC6 and HDAC10 reduced angiogenesis-associated VEGFR1 and VEGFR2 [
117]. Many reports showed up- or downregulation of SIRT genes in tumors. Along this line, SIRT1 was upregulated in lung cancer [
118], prostate cancer [
119], and leukemia [
59], and downregulated in colon tumors [
120]. Embryonic lethality, ascribed to reduced ability to repair DNA damage, was observed in mice lacking SIRT1 [
121,
122,
123]. High levels of SIRT1 in AR-positive prostate cancer cell lines repress their multiplication. Indeed, SIRT1 can elicit senescence and avoid tumorigenesis [
124]. SIRT2 is frequently downregulated in human gliomas [
124,
125,
126].
3.1. HDAC in Different Cancer Stages
3.1.1. Cell Cycle Progression and Apoptosis
Several HDACs (from 1 to 6) are involved in tumor development, and their loss promotes cell proliferation dysregulation [
113,
116,
127,
128,
129]. HDAC1 reduces suppressors of the cell cycle mutually with Rb and by altering E2F1 activity [
130]. The inhibition of HDAC 1-2 induces cell cycle arrest [
131]. HDAC1 is also involved in G
1/S and G
2/M transitions. Another study also showed that HDAC1 knock-down contributes to G
2/M phase arrest [
132]. Similarly, both HDAC3 and HDAC10 modulate the G
2/M transition [
133,
134]. High levels of Sp1 due to HDAC1/2/6 activities promote the division of cancer cells and G
2/M progression [
135]. Knockdown of HDAC3 induced gathering of cells at the G
2/M phase [
103], whereas in osteosarcoma cells, this effect causes siRNA-mediated HDAC1 depletion [
129].
Cell cycle interruption at the G
2/M stage in renal cancer, following the inhibition of HDAC6 and HDAC3, has been ascribed to proteasomal alteration of Aurora B and A [
136]. Furthermore, SIRT1 can suppress the cell cycle through the blockage of p53-dependent pathways [
137]. HDAC11 negatively affects E2F7, E2F8, and cell cycle suppressors, leading to survival of tumor cells [
138]. Moreover, HDACs act as apoptosis regulators, as the interruption of this process is a critical factor for tumor progression and, therefore, is considered a hallmark of tumor progression. HDACs contribute to the extrinsic, as well as intrinsic, apoptotic pathways. Regarding the extrinsic apoptotic pathways, HDACs can obstruct TRAIL or TGF-b-mediated pathways, while pro- and antiapoptotic factors are altered in the intrinsic pathway [
139].
3.1.2. Differentiation
During differentiation, establishing a specific gene expression profile is harmonized by epigenetic modifications, e.g., histone acetylation. In this context, HDAC3 was recruited by RARPML [
140], while HDAC4 interacted with RAR-PLZF [
141] to repress differentiation-specific transcription. A close mechanism of retinoic acid signaling limitation in hematopoietic cells was recorded in AML1-ETO fusion proteins, which bind to HDAC1, 2, and 3 [
142,
143]. HDAC8 is a key regulator of cancer cell differentiation [
144], and HDAC8 overexpression is associated with neuroblastoma progression.
3.1.3. DNA Damage Response
HDACs contribute to DNA damage repair (DDR) responses via their key role in remodeling chromatin and regulating the acetylation patterns of proteins associated with DNA [
145]. The inhibition of HDAC blocks double-strand break (DSB) repair and radio-sensitizes cancerous cells. In this line, HDAC2 and HDAC1 bind to DNA damage regions, to deacetylate histones at H3K56 and H4K16 and promote non-homologous end-joining pathways, which accelerate DSB repair [
146]. Moreover, HDAC3 is involved in nucleotide excision repair (NER) [
147], whereas HDAC9 and HDAC10 contribute to homologous recombination [
148,
149]. HDAC6, in association with DNA mismatch repair protein (MSH2), acts as an MSH2 inhibitor through deacetylation and ubiquitination [
150]. In addition, Sirtuins interact with numerous proteins regulating several DDR pathways [
151]. In tumor cells, SIRT1 restraints p53 acetylation, contributing to cell survival [
148,
152]. SIRT6 phosphorylation is directly engaged in DNA damage sites to promote DSB repair [
153,
154]. In leukemia-initiating cells, the inhibition of SIRT6 or HDAC8 engenders a DNA repair deficit in homologous recombination and the NHEJ pathway [
155].
3.1.4. Metastasis
The capacity to disperse and metastasize represents the deadliest signature of tumor cells. Numerous works have evidenced that HDACs regulate metastasis in various cancers. An important player in metastasis is the transition from adherent epithelial cells to motile mesenchymal cells capable of leaving the primary tumor site. In embryonic development, EMT is a crucial path for cell migration during gastrulation [
156]. In colorectal cancer, HDAC3 was engaged to Runx 2 promoter and hampered metastasis [
157]. Research findings showed that HDAC7 enhances EphA2 expression by downregulating miR-4465 expression, positively affecting tumor proliferation, spread, and invasion in nasopharyngeal cancer [
158]. Similarly, HDAC11 leads to the overexpression of RRM2, a gene involved in promigratory and metastatic phenotypes [
138]. In prostate cancer, ZEB1 and SIRT1 together bind to CDH1, promoting metastasis [
159], while SIRT1 elicited EMT through Fra-1 over-expression in colorectal cancer [
160].
3.1.5. Angiogenesis
Angiogenesis involves the creation and addition of new blood vessels and is pivotal for the development of tumors [
166,
167]. The first steps of angiogenesis are elicited by hypoxia or a hypoxic microenvironment, while its advancement is mainly controlled by hypoxia-inducible factor 1a (HIF-1a). Overall, HDACs monitor the balance between pro- and anti-angiogenic proteins. In this context, HDAC inhibition exerts anti-angiogenic activity via inhibition of pro-angiogenic gene expression. Under hypoxia conditions, Class I HDACs, mRNA, and protein were overexpressed in vitro in primary and malignant cells [
168]. HDAC1 deacetylates HIF-1a, contributing to preventing HIF-1a loss. On the other hand, dysregulated levels of HDAC1 lead to high levels of HIF-1a and VEGF in tumors, which in turn enhances angiogenesis [
169]. HDAC4, 6, 10, and SIRTs display similar pathways [
139], whereas HDAC4, 5, and 6, acting as mediators of HIF-1 activity, require cofactors (HSP90 and p300) [
170]. On the contrary, it was shown that SIRT1 deacetylated HIF-1a, which reduces the interaction of HIF-1a with p300, which reduces HIF-1a activity. In endothelial cells, HDAC5 reduced the expression of pro-angiogenic genes (FGF2 and Slit2) [
171]. Additionally, HDAC5 represses cysteine-rich angiogenic inducer 61 (CYR-61), a well-known antifibrotic and pro-angiogenic mediator, inhibiting angiogenesis [
172]. HDAC6 enhances angiogenesis via deacetylation of cortactin, an actin-remodeling protein [
173].
3.1.6. Autophagy
Autophagy is a process that suppresses damaged subcellular fractions, helping to intercept the transformation of normal cells to cancerous ones [
174,
175]. Published data showed that Class I HDACs mediate autophagic flux in mice [
50], whereas elimination of HDAC1 and HDAC2 impedes autophagic flux [
176]. On the other hand, HDAC4 and HDAC5 influence autophagic flux by acting as positive regulators of tumor cell development. HDAC6 promotes autophagy, based on its connection with microtubule proteins [
177,
178]. In this regard, autophagy might be actively enhanced and play a compensatory role for HDAC6 under ubiquitin-proteasome system damage [
120,
148]. At the same time, HDAC6 shows a significant role in ubiquitin-selective quality control autophagy, instead of starvation-induced autophagy [
163]. Similarly, Parkin-mediated mitochondrial ubiquitination could engage the autophagic actors, i.e., HDAC6 and p62 [
179]. HDAC10 knock-down induces autophagosome/lysosome fusion blockade and restriction of autophagic flux, which sensitizes cells to chemotherapy [
180]. HDAC10 also deacetylates HSP70 protein members associated with autophagy-mediated cell longevity [
180]. In contrast, SIRT1 displays a dual role in autophagy [
181], where it is necessary to trigger starvation-induced autophagy [
127,
182]. Moreover, SIRT1 deacetylates forkhead box O3 (FOXO3), leading to proteasomal degradation and, thus, contributing to the overexpression of numerous autophagic genes.
In embryonic stem cells (ESCs), SIRT1 affects the PI3K/Beclin 1 and mTOR pathways, affecting oxidative stress-induced autophagy [
183]. SIRT2 detaches from FOXO1 under stress conditions, which promotes hyperacetylated FOXO1, promoting the autophagic process [
184]. In contrast, deacetylation of lactate dehydrogenase B (LDHB) by SIRT5 intensifies its effect. Protons (H
+) generated by LDHB promote autophagy in tumor cells [
181,
185]. Furthermore, SIRT5 is involved in ammonia-induced autophagy, via glutamine metabolism remodeling [
184]. SIRT6 promotes autophagy by hampering the transcriptional repressor Nkx3.2, resulting in the expression of GATA5 [
186].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27082568