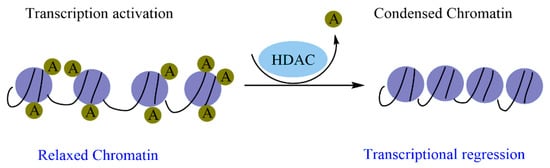
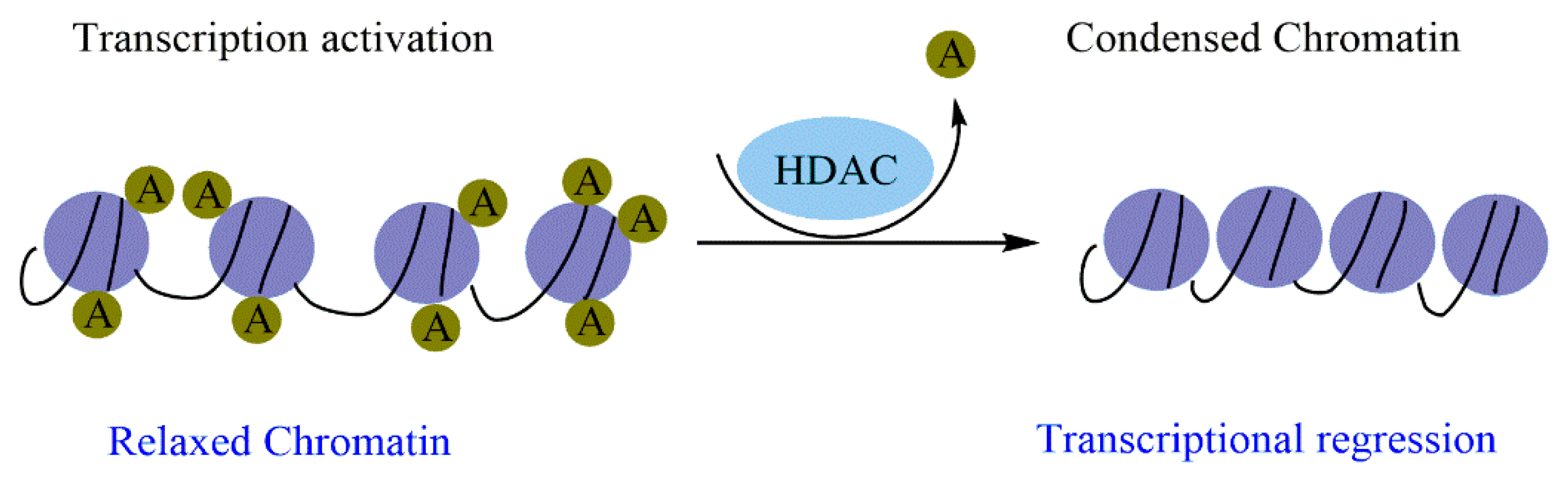
Histone modification is an essential mark in maintaining cellular memory and, therefore, loss of this mark can lead to tumor transformation. As these epigenetic changes are reversible, the use of molecules that can restore the functions of the enzymes responsible for the changes is therapeutically necessary. Natural molecules, mainly those isolated from medicinal plants, have demonstrated significant inhibitory properties against enzymes related to histone modifications, particularly histone deacetylases (HDACs). Flavonoids, terpenoids, phenolic acids, and alkaloids exert significant inhibitory effects against HDAC and exhibit promising epi-drug properties. This suggests that epi-drugs against HDAC could prevent and treat various human cancers.
- epigenetic
- histone deacetylases
- cancer
- natural compounds