 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlo Smirne | + 10506 word(s) | 10506 | 2022-02-08 09:16:42 | | | |
| 2 | Bruce Ren | -1 word(s) | 10505 | 2022-02-14 01:56:24 | | | | |
| 3 | Bruce Ren | -285 word(s) | 10220 | 2022-02-14 02:07:32 | | | | |
| 4 | Bruce Ren | -285 word(s) | 10220 | 2022-02-14 02:09:02 | | | | |
| 5 | Bruce Ren | -285 word(s) | 10220 | 2022-02-14 02:11:09 | | |
Video Upload Options
Non-alcoholic fatty liver disease (NAFLD) is a challenging disease caused by multiple factors, which may partly explain why it still remains an orphan of adequate therapies. It highlights the interaction between oxidative stress (OS) and disturbed lipid metabolism. Several reactive oxygen species generators, including those produced in the gastrointestinal tract, contribute to the lipotoxic hepatic (and extrahepatic) damage by fatty acids and a great variety of their biologically active metabolites in a “multiple parallel-hit model”. This leads to inflammation and fibrogenesis and contributes to NAFLD progression. The alterations of the oxidant/antioxidant balance affect also metabolism-related organelles, leading to lipid peroxidation, mitochondrial dysfunction, and endoplasmic reticulum stress. This OS-induced damage is at least partially counteracted by the physiological antioxidant response. Therefore, modulation of this defense system emerges as an interesting target to prevent NAFLD development and progression. For instance, probiotics, prebiotics, diet, and fecal microbiota transplantation represent new therapeutic approaches targeting the gut microbiota dysbiosis. The OS and its counter-regulation are under the influence of individual genetic and epigenetic factors as well.
1. Introduction
2. Evidences on the Role of Oxidative Stress on NAFLD Progression
2.1. Markers of Immunological Responses to Inflammation and Oxidative Stress during NAFLD Evolution
2.2. Mitochondrial Oxidative Injury as a Key Pathway That Links Saturated Fat Intake to the Development and Progression of NAFLD
2.2.1. Mitochondrial Dysfunction and Oxidative Stress in NAFLD/NASH
2.2.2. Diagnostic Tools for Mitochondrial Dysfunction in NAFLD
2.3. Gut Microbiota and Oxidative Stress in NAFLD, with Possible Therapeutic Implications
2.3.1. Role of Microbiota in Hepatic Steatosis and Oxidative Stress
2.3.2. Modulation of Gut-Liver Axis: Effects on Oxidative Stress
2.4. Relationship of Oxidative Stress and the Development and/or Progression of NAFLD-HCC
2.5. Biomarkers of Oxidative Stress in NAFLD/NASH
| Oxidative Stress Markers | Disease Status | Experimental Model and Species |
Sample | Changes in Concentration/ Activity/Expression |
Reference(s) |
|---|---|---|---|---|---|
| TBARS/MDA | Steatosis, NASH | HFD, HF-HSD, MCD, ob/ob mice, CDHF diet | Liver | ↑ | [251][252] |
| HCD | ↓ | ||||
| HFD, MCD | NS | ||||
| 4-HNE | Steatosis, NASH | HFD, MCD, HF-HSD, CDHF diet | Liver | ↑ | [253][254] |
| 8-OH-dG | NASH | MCD, ob/ob mice HFMCD | Liver | ↑ ↓ |
[255] |
| 8-isoprostane | NASH | HFMCD | Liver | ↑ | [256] |
| Protein carbonyl | Steatosis, NASH | HFD, MCD HFD |
Liver | ↑ NS |
[257] |
| Nitrotyrosine | NASH | HFD, ob/ob mice | Liver | ↑ | [258] |
| Periostin | NASH | HFD, ob/ob mice | Serum, liver | ↑ | [259] |
| CYP2E1 | Steatosis, NASH | HFD, HF-HSD, CDHF | Liver | ↑ | [252][260] |
| Dityrosine | Steatosis | HFD | Liver | ↑ | [261] |
| Hydroxyproline | NASH | MCD/WD | Liver | ↑ | [262] |
| H2O2 | NASH | MCD | Liver | ↑ | [250] |
| Lipid peroxide | NASH | MCD | Liver | ↑ | [263] |
| NADPH oxidase | NASH | ob/ob mice | Liver | ↑ | [264] |
| Xanthine oxidase | Steatosis | HFD | Liver | ↑ | [265] |
| Oxidative Stress Markers | Disease Status | Sample | Changes in Concentration/ Activity/Expression |
Reference(s) |
|---|---|---|---|---|
| TBARS/MDA | Steatosis, NASH | Serum, liver, blood | ↑ | [269][53][270] |
| Serum | NS | |||
| 4-HNE | NASH | Liver | ↑ | [271] |
| Hydroperoxides | NASH | Liver | ↑ | [272] |
| 8-OH-dG | Steatosis, NASH | Liver, plasma Liver |
↑ NS |
[273] |
| 8-isoprostane | NASH | Plasma | NS | [267] |
| Protein carbonyl | Steatosis, NASH | Liver | ↑ | [268] |
| Nitrotyrosine | Steatosis, NASH | Liver | ↑ | [25] |
| Blood | NS | |||
| Periostin | Steatosis, NASH | Serum, plasma, liver Serum, plasma |
↑ | [274][275][276][277] |
| NS | ||||
| Nitric oxide | Steatosis, NASH | Serum, blood | ↑ | [278][279] |
| CYP2E1 | Steatosis, NASH | Liver | ↑ | [280] |
| NS |
| Antioxidant Marker | Disease Status | Experimental Model and Species |
Sample | Changes in Concentration/ Activity/Expression |
Reference(s) |
|---|---|---|---|---|---|
| SOD | Steatosis, NASH | HF, HFD, OLETF rats, MCD | Liver | ↓ | [286][288] |
| HFD, MCD | ↑ | ||||
| HFD | NS | ||||
| Catalase | Steatosis, NASH | HFD, MCD, HCD | Liver | ↓ | [53][285] |
| MCD | ↑ | ||||
| GPx | Steatosis, NASH | HFD, MCD, HF | Liver | ↓ | [267][268] |
| MCD | ↑ | ||||
| HFD | NS | ||||
| GSH | Steatosis, NASH | HFD, HCD, MCD, ob/ob mice | Liver | ↓ | [288] |
| OLETF rats, HF MCD | ↑ | ||||
| GR | NASH | MCD | Liver | ↓ | [282] |
| Antioxidant Marker | Disease Status | Sample | Changes in Concentration/ Activity/Expression |
Reference(s) |
|---|---|---|---|---|
| SOD | Steatosis, NASH | Serum, plasma, liver | ↓ | [286][288] |
| Blood, serum | ↑ | |||
| Serum | NS | |||
| Catalase | Steatosis, NASH | Plasma, blood, liver | ↓ | [53][285] |
| Serum | NS | |||
| GPx | Steatosis, NASH | Liver, serum | ↓ | [267][268] |
| Blood | ↑ | |||
| Serum | NS | |||
| GSH | Steatosis, NASH | Liver, blood | ↓ | [288] |
| Serum | ↑ | |||
| GR | Steatosis, NASH | Serum, blood | ↑ | [286] |
| TRX | Steatosis | Serum | ↑ | [287] |
| α-Tocopherol | Steatosis, NASH | Serum | ↓ | [275] |
| NS | ||||
| Ubiquinone | Steatosis | Serum | ↓ | [289] |
| Bilirubin | Steatosis, NASH | Serum | ↓ | [279] |
| Ascorbic acid | Steatosis, NASH | Serum | ↓ | [288] |
| NS |
3. Conclusions
References
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78.
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575.
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555.
- Khan, F.Z.; Perumpail, R.B.; Wong, R.J.; Ahmed, A. Advances in hepatocellular carcinoma: Nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2155–2161.
- Hamaguchi, M.; Kojima, T.; Itoh, Y.; Harano, Y.; Fujii, K.; Nakajima, T.; Kato, T.; Takeda, N.; Okuda, J.; Ida, K.; et al. The severity of ultrasonographic findings in nonalcoholic fatty liver disease reflects the metabolic syndrome and visceral fat accumulation. Am. J. Gastroenterol. 2007, 102, 2708–2715.
- Shenoy-Bhangle, A.; Baliyan, V.; Kordbacheh, H.; Guimaraes, A.R.; Kambadakone, A. Diffusion weighted magnetic resonance imaging of liver: Principles, clinical applications and recent updates. World J. Hepatol. 2017, 9, 1081–1091.
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321.
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728.
- Noureddin, M.; Mato, J.M.; Lu, S.C. Nonalcoholic fatty liver disease: Update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp. Biol. Med. 2015, 240, 809–820.
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845.
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778.
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351.
- Targher, G.; Marchesini, G.; Byrne, C.D. Risk of type 2 diabetes in patients with non-alcoholic fatty liver disease: Causal association or epiphenomenon? Diabetes Metab. 2016, 42, 142–156.
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944.
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A meta-analysis. Medicine 2017, 96, e8179.
- Tesfay, M.; Goldkamp, W.J.; Neuschwander-Tetri, B.A. NASH: The Emerging Most Common Form of Chronic Liver Disease. Mo. Med. 2018, 115, 225.
- Polimeni, L.; del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336.
- Vergara, D.; Casadei-Gardini, A.; Giudetti, A.M. Oxidative Molecular Mechanisms Underlying Liver Diseases: From Systems Biology to the Personalized Medicine. Oxid. Med. Cell. Longev. 2019, 2019, 7864316.
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231.
- Hruszkewycz, A.M. Evidence for mitochondrial DNA damage by lipid peroxidation. Biochem. Biophys. Res. Commun. 1988, 153, 191–197.
- Chen, J.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim. Biophys. Acta 1998, 1380, 336–344.
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192.
- Gambino, R.; Musso, G.; Cassader, M. Redox balance in the pathogenesis of nonalcoholic fatty liver disease: Mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1325–1365.
- Sastre, J.; Pallardó, F.V.; Llopis, J.; Furukawa, T.; Vinã, J.R.; Viña, J. Glutathione depletion by hyperphagia-induced obesity. Life Sci. 1989, 45, 183–187.
- Strauss, R.S.; Barlow, S.E.; Dietz, W.H. Prevelance of abnormal serum aminotransferase values in overweight and obese adolescents. J. Pediatr. 2000, 136, 727–733.
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912.
- George, D.K.; Goldwurm, S.; Macdonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998, 114, 311–318.
- Chitturi, S.; Weltman, M.; Farrell, G.C.; McDonald, D.; Liddle, C.; Samarasinghe, D.; Lin, R.; Abeygunasekera, S.; George, J. HFE mutations, hepatic iron, and fibrosis: Ethnic-specific association of NASH with C282Y but not with fibrotic severity. Hepatology 2002, 36, 142–149.
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St. Pierre, T.G.; Webb, M.; Ching, H.L.I.; Kava, J.; Bynevelt, M.; Macquillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015, 61, 1555–1564.
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794.
- Park, S.H.; Lee, W.Y.; Lee, Y.S.; Rhee, E.J.; Kim, S.W. The relative effects of obesity and insulin resistance on cardiovascular risk factors in nondiabetic and normotensive men. Korean J. Intern. Med. 2004, 19, 75–80.
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793.
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279.
- Dongiovanni, P.; Romeo, S.; Valenti, L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. Biomed Res. Int. 2015, 2015, 460190.
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273.
- Wigg, A.J.; Roberts-Thomson, I.C.; Grose, R.H.; Cummins, A.G.; Dymock, R.B.; McCarthy, P.J. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211.
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887.
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347.
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 38–42.
- Neish, A.S. Microbes in gastrointestinal health and disease. Gastroenterology 2009, 136, 65–80.
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7.
- Petrasek, J.; Dolganiuc, A.; Csak, T.; Kurtjones, E.A.; Szabo, G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology 2011, 140, 697–708.e4.
- Petrasek, J.; Csak, T.; Ganz, M.; Szabo, G. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2013, 28, 93.
- Konerman, M.A.; Jones, J.C.; Harrison, S.A. Pharmacotherapy for NASH: Current and emerging. J. Hepatol. 2018, 68, 362–375.
- Sanchez-Valle, V.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860.
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174.
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2019, 31, 189–206.e8.
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384.
- Shepard, C.R. TLR9 in MAFLD and NASH: At the Intersection of Inflammation and Metabolism. Front. Endocrinol. 2021, 11, 613639.
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746.
- Ouyang, X.; Han, S.N.; Zhang, J.Y.; Nemeth, B.T.; Pacher, P.; Feng, D.; Bataller, R.; Cabezas, J.; Stärkel, P.; Caballeria, J.; et al. Digoxin Suppresses Pyruvate Kinase M2-Promoted HIF-1α Transactivation in Steatohepatitis. Cell Metab. 2018, 27, 339–350.e3.
- Peters, K.M.; Wilson, R.B.; Borradaile, N.M. Non-parenchymal hepatic cell lipotoxicity and the coordinated progression of non-alcoholic fatty liver disease and atherosclerosis. Curr. Opin. Lipidol. 2018, 29, 417–422.
- Bataller, R.; Schwabe, R.F.; Choi, Y.H.; Yang, L.; Paik, Y.H.; Lindquist, J.; Qian, T.; Schoonhoven, R.; Hagedorn, C.H.; Lemasters, J.J.; et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Investig. 2003, 112, 1383–1394.
- Paik, Y.H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Österreicher, C.H.; Kisseleva, T.; Brenner, D.A. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011, 53, 1730–1741.
- Thuy, L.T.T.; Hai, H.; Kawada, N. Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis. Clin. Mol. Hepatol. 2020, 26, 280.
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291.
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. Innov. Hepatol. 2021, 3, 100301.
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-kappaB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243.
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858.
- Komeili-Movahhed, T.; Bassirian, M.; Changizi, Z.; Moslehi, A. SIRT1/NFκB pathway mediates anti-inflammatory and anti-apoptotic effects of rosmarinic acid on in a mouse model of nonalcoholic steatohepatitis (NASH). J. Recept. Signal Transduct. Res. 2021, 1–10.
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760.
- Solano-Urrusquieta, A.; Morales-González, J.A.; Castro-Narro, G.E.; Cerda-Reyes, E.; Flores-Rangel, P.D.; Fierros-Oceguera, R. NRF-2 and nonalcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 458–465.
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 367–398.
- Gardner, H.W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989, 7, 65–86.
- Meakin, P.J.; Chowdhry, S.; Sharma, R.S.; Ashford, F.B.; Walsh, S.V.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; Dillon, J.F.; Hayes, J.D.; Ashford, M.L.J. Susceptibility of Nrf2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol. Cell. Biol. 2014, 34, 3305–3320.
- Mohs, A.; Otto, T.; Schneider, K.M.; Peltzer, M.; Boekschoten, M.; Holland, C.H.; Hudert, C.A.; Kalveram, L.; Wiegand, S.; Saez-Rodriguez, J.; et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J. Hepatol. 2021, 74, 638–648.
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp. Cell Res. 2021, 405, 112689.
- Das, D.; Paul, A.; Lahiri, A.; Adak, M.; Maity, S.K.; Sarkar, A.; Paul, S.; Chakrabarti, P. Proteasome dysfunction under compromised redox metabolism dictates liver injury in NASH through ASK1/PPARγ binodal complementary modules. Redox Biol. 2021, 45, 102043.
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727.
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290.
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G891–G899.
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564.
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304.
- Li, D.Y.; Xiong, X.Z. ICOS + Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104.
- Tang, G.; Qin, Q.; Zhang, P.; Wang, G.; Liu, M.; Ding, Q.; Qin, Y.; Shen, Q. Reverse signaling using an inducible costimulator to enhance immunogenic function of dendritic cells. Cell. Mol. Life Sci. 2009, 66, 3067–3080.
- Occhipinti, S.; Dianzani, C.; Chiocchetti, A.; Boggio, E.; Clemente, N.; Gigliotti, C.L.; Soluri, M.F.; Minelli, R.; Fantozzi, R.; Yagi, J.; et al. Triggering of B7h by the ICOS modulates maturation and migration of monocyte-derived dendritic cells. J. Immunol. 2013, 190, 1125–1134.
- Gigliotti, C.L.; Boggio, E.; Clemente, N.; Shivakumar, Y.; Toth, E.; Sblattero, D.; D’Amelio, P.; Isaia, G.C.; Dianzani, C.; Yagi, J.; et al. ICOS-Ligand Triggering Impairs Osteoclast Differentiation and Function In Vitro and In Vivo. J. Immunol. 2016, 197, 3905–3916.
- Koh, K.H.; Cao, Y.; Mangos, S.; Tardi, N.J.; Dande, R.R.; Lee, H.W.; Samelko, B.; Altintas, M.M.; Schmitz, V.P.; Lee, H.; et al. Nonimmune cell-derived ICOS ligand functions as a renoprotective αvβ3 integrin-selective antagonist. J. Clin. Investig. 2019, 129, 1713–1726.
- Ramavath, N.N.; Gadipudiaik, L.I.; Provera, A.; Gigliotti, L.C.; Boggio, E.; Clemente, N.; Dianzani, U.; Albano, E.; Sutti, S. Co-stimulatory signals mediated by ICOS/ICOS-L dyad influence the differentiation of liver NASH-associated macrophages (NAMs). In Abstracts of The International Liver Congress™ 2021, Proceedings of the International Liver Congress (ILC), Digital, 23–26 June 2021. J. Hepatol. 2021, 75, S598.
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199.
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742.
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411.
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic. Biol. Med. 2008, 44, 1259–1272.
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99.
- Meex, R.C.R.; Blaak, E.E. Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD. Mol. Nutr. Food Res. 2021, 65, e1900942.
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507.
- Satapati, S.; Kucejova, B.; Duarte, J.A.G.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Arya Nair, L.; Livingston, K.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462.
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092.
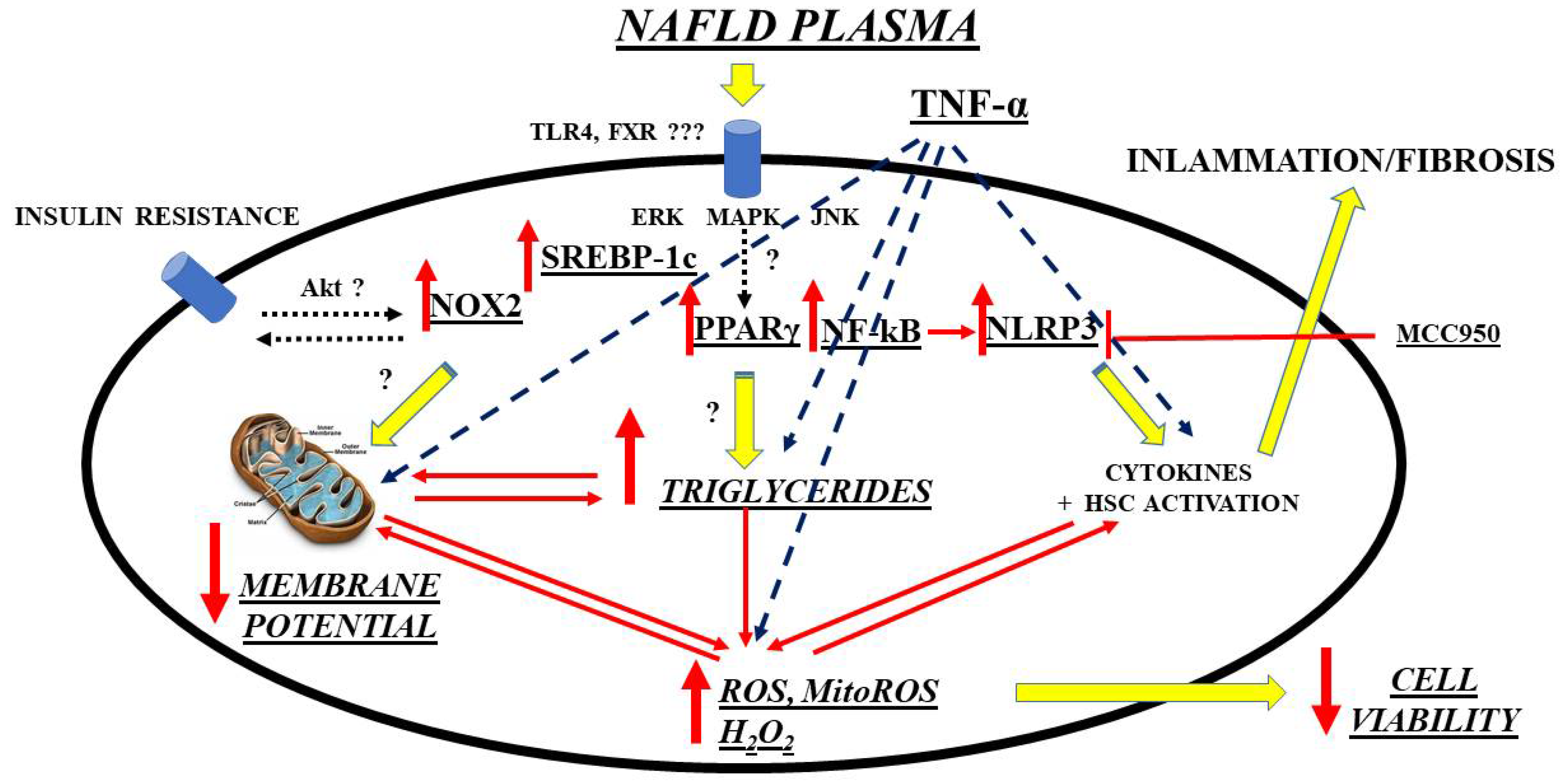
- Grossini, E.; Garhwal, D.P.; Calamita, G.; Romito, R.; Rigamonti, C.; Minisini, R.; Smirne, C.; Surico, D.; Bellan, M.; Pirisi, M. Exposure to Plasma from Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 2021, 8, 693997.
- Karkucinska-Wieckowska, A.; Simoes, I.C.M.; Kalinowski, P.; Lebiedzinska-Arciszewska, M.; Zieniewicz, K.; Milkiewicz, P.; Górska-Ponikowska, M.; Pinton, P.; Malik, A.N.; Krawczyk, M.; et al. Mitochondria, oxidative stress and nonalcoholic fatty liver disease: A complex relationship. Eur. J. Clin. Investig. 2021, e13622.
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated with Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728.
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880.
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262.
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141.
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218.
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438.
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Willem Greve, J.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986.
- Sullivan, E.M.; Fix, A.; Crouch, M.J.; Sparagna, G.C.; Zeczycki, T.N.; Brown, D.A.; Shaikh, S.R. Murine diet-induced obesity remodels cardiac and liver mitochondrial phospholipid acyl chains with differential effects on respiratory enzyme activity. J. Nutr. Biochem. 2017, 45, 94–103.
- Han, X.; Yang, J.; Yang, K.; Zhongdan, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428.
- Di Ciaula, A.; Calamita, G.; Shanmugam, H.; Khalil, M.; Bonfrate, L.; Wang, D.Q.H.; Baffy, G.; Portincasa, P. Mitochondria Matter: Systemic Aspects of Nonalcoholic Fatty Liver Disease (NAFLD) and Diagnostic Assessment of Liver Function by Stable Isotope Dynamic Breath Tests. Int. J. Mol. Sci. 2021, 22, 7702.
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198.
- Afolabi, P.; Wright, M.; Wootton, S.A.; Jackson, A.A. Clinical utility of 13C-liver-function breath tests for assessment of hepatic function. Dig. Dis. Sci. 2013, 58, 33–41.
- Gorowska-Kowolik, K.; Chobot, A.; Kwiecien, J. 13 C Methacetin Breath Test for Assessment of Microsomal Liver Function: Methodology and Clinical Application. Gastroenterol. Res. Pract. 2017, 2017, 7397840.
- Afolabi, P.R.; Scorletti, E.; Smith, D.E.; Almehmadi, A.A.; Calder, P.C.; Byrne, C.D. The characterisation of hepatic mitochondrial function in patients with non-alcoholic fatty liver disease (NAFLD) using the 13 C-ketoisocaproate breath test. J. Breath Res. 2018, 12, 046002.
- Campo, L.; Eiseler, S.; Apfel, T.; Pyrsopoulos, N. Fatty Liver Disease and Gut Microbiota: A Comprehensive Update. J. Clin. Transl. Hepatol. 2019, 7, 56–60.
- Lau, E.; Carvalho, D.; Freitas, P. Gut Microbiota: Association with NAFLD and Metabolic Disturbances. Biomed Res. Int. 2015, 2015, 979515.
- Zhou, J.; Tripathi, M.; Sinha, R.A.; Singh, B.K.; Yen, P.M. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res. 2021, 7, 11.
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772.
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984.
- Burz, S.D.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.M.; Chêne, L.; Blottière, H.M.; Gérard, P. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 2021, 9, 199.
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371.
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7.
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G.; et al. Donor Fecal Microbiota Transplantation Alters Gut Microbiota and Metabolites in Obese Individuals With Steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590.
- Hrncir, T.; Hrncirova, L.; Kverka, M.; Hromadka, R.; Machova, V.; Trckova, E.; Kostovcikova, K.; Kralickova, P.; Krejsek, J.; Tlaskalova-Hogenova, H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms 2021, 9, 957.
- Jennison, E.; Byrne, C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 22–43.
- Bakhshimoghaddam, F.; Alizadeh, M. Contribution of gut microbiota to nonalcoholic fatty liver disease: Pathways of mechanisms. Clin. Nutr. ESPEN 2021, 44, 61–68.
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; De Maria, S.; Cartenì, M.; Nardone, G. Gut—Liver axis: The impact of gut microbiota on non alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476.
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228.
- Nicoletti, A.; Ponziani, F.R.; Biolato, M.; Valenza, V.; Marrone, G.; Sganga, G.; Gasbarrini, A.; Miele, L.; Grieco, A. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 2019, 25, 4814–4834.
- Auguet, T.; Bertran, L.; Binetti, J. Intestinal Dysbiosis and Non-Alcoholic Fatty Liver Disease. In Human Microbiome; Beloborodova, N.V., Grechko, A.V., Eds.; Intech Open Limited: London, UK, 2020; pp. 1–24.
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023.
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65.
- Jasirwan, O.M.C.; Muradi, A.; Hasan, I.; Simadibrata, M.; Rinaldi, I. Correlation of gut Firmicutes/Bacteroidetes ratio with fibrosis and steatosis stratified by body mass index in patients with non-alcoholic fatty liver disease. Biosci. Microbiota Food Health 2021, 40, 50–58.
- Zhu, L.; Baker, R.D.; Baker, S.S. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr. Res. 2015, 77, 245–251.
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712.
- Eswaran, S.; Babbar, A.; Drescher, H.K.; Hitch, T.C.A.; Clavel, T.; Muschaweck, M.; Ritz, T.; Kroy, D.C.; Trautwein, C.; Wagner, N.; et al. Upregulation of Anti-Oxidative Stress Response Improves Metabolic Changes in L-Selectin-Deficient Mice but Does Not Prevent NAFLD Progression or Fecal Microbiota Shifts. Int. J. Mol. Sci. 2021, 22, 7314.
- Wang, W.; Li, Q.; Chai, W.; Sun, C.; Zhang, T.; Zhao, C.; Yuan, Y.; Wang, X.; Liu, H.; Ye, H. Lactobacillus paracasei Jlus66 extenuate oxidative stress and inflammation via regulation of intestinal flora in rats with non alcoholic fatty liver disease. Food Sci. Nutr. 2019, 7, 2636–2646.
- Park, E.; Jeong, J.J.; Won, S.M.; Sharma, S.P.; Gebru, Y.A.; Ganesan, R.; Gupta, H.; Suk, K.T.; Kim, D.J. Gut Microbiota-Related Cellular and Molecular Mechanisms in the Progression of Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2634.
- Shen, J.; Obin, M.S.; Zhao, L. The gut microbiota, obesity and insulin resistance. Mol. Asp. Med. 2013, 34, 39–58.
- Diamant, M.; Blaak, E.E.; de Vos, W.M. Do nutrient-gut-microbiota interactions play a role in human obesity, insulin resistance and type 2 diabetes? Obes. Rev. 2011, 12, 272–281.
- Cassard, A.-M.; Gérard, P.; Perlemuter, G. Microbiota, Liver Diseases, and Alcohol. Microbiol. Spectr. 2017, 5.
- Kullisaar, T.; Songisepp, E.; Mikelsaar, M.; Zilmer, K.; Vihalemm, T.; Zilmer, M. Antioxidative probiotic fermented goats’ milk decreases oxidative stress-mediated atherogenicity in human subjects. Br. J. Nutr. 2003, 90, 449–456.
- Arora, T.; Singh, S.; Sharma, R.K. Probiotics: Interaction with gut microbiome and antiobesity potential. Nutrition 2013, 29, 591–596.
- Park, D.Y.; Ahn, Y.T.; Huh, C.S.; Mcgregor, R.A.; Choi, M.S. Dual probiotic strains suppress high fructose-induced metabolic syndrome. World J. Gastroenterol. 2013, 19, 274–283.
- Castillo, V.; Figueroa, F.; González-Pizarro, K.; Jopia, P.; Ibacache-Quiroga, C. Probiotics and Prebiotics as a Strategy for Non-Alcoholic Fatty Liver Disease, a Narrative Review. Foods 2021, 10, 1719.
- Moszak, M.; Szulińska, M.; Walczak-Gałęzewska, M.; Bogdański, P. Nutritional Approach Targeting Gut Microbiota in NAFLD-To Date. Int. J. Environ. Res. Public Health 2021, 18, 1616.
- Nagashimada, M.; Honda, M. Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. Int. J. Mol. Sci. 2021, 22, 8008.
- Khan, A.; Ding, Z.; Ishaq, M.; Bacha, A.S.; Khan, I.; Hanif, A.; Li, W.; Guo, X. Understanding the Effects of Gut Microbiota Dysbiosis on Nonalcoholic Fatty Liver Disease and the Possible Probiotics Role: Recent Updates. Int. J. Biol. Sci. 2021, 17, 818–833.
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702.
- Porras, D.; Nistal, E.; Martínez-Flórez, S.; González-Gallego, J.; García-Mediavilla, M.V.; Sánchez-Campos, S. Intestinal Microbiota Modulation in Obesity-Related Non-alcoholic Fatty Liver Disease. Front. Physiol. 2018, 9, 1813.
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264.
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075.
- Jegatheesan, P.; De Bandt, J.P. Fructose and NAFLD: The Multifaceted Aspects of Fructose Metabolism. Nutrients 2017, 9, 230.
- Roeb, E.; Weiskirchen, R. Fructose and Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2021, 12, 634344.
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999.
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159.
- Xie, C.; Halegoua-Demarzio, D. Role of Probiotics in Non-alcoholic Fatty Liver Disease: Does Gut Microbiota Matter? Nutrients 2019, 11, 2837.
- Nabavi, S.; Rafraf, M.; Somi, M.H.; Homayouni-Rad, A.; Asghari-Jafarabadi, M. Effects of probiotic yogurt consumption on metabolic factors in individuals with nonalcoholic fatty liver disease. J. Dairy Sci. 2014, 97, 7386–7393.
- Famouri, F.; Shariat, Z.; Hashemipour, M.; Keikha, M.; Kelishadi, R. Effects of Probiotics on Nonalcoholic Fatty Liver Disease in Obese Children and Adolescents. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 413–417.
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 5688.
- Javadi, L.; Khoshbaten, M.; Safaiyan, A.; Ghavami, M.; Abbasi, M.M.; Gargari, B.P. Pro- and prebiotic effects on oxidative stress and inflammatory markers in non-alcoholic fatty liver disease. Asia Pac. J. Clin. Nutr. 2018, 27, 1031–1039.
- Lei, K.; Li, Y.L.; Wang, Y.; Wen, J.; Wu, H.Z.; Yu, D.Y.; Li, W.F. Effect of dietary supplementation of Bacillus subtilis B10 on biochemical and molecular parameters in the serum and liver of high-fat diet-induced obese mice. J. Zhejiang Univ. Sci. B 2015, 16, 487–495.
- Kozmus, C.E.P.; Moura, E.; Serrão, M.P.; Real, H.; Guimarães, J.T.; Guedes-de-Pinho, P.; Duarte, B.P.; Marques, F.; Martins, M.J.; Vieira-Coelho, M.A. Influence of dietary supplementation with dextrin or oligofructose on the hepatic redox balance in rats. Mol. Nutr. Food Res. 2011, 55, 1735–1739.
- Ekhlasi, G.; Zarrati, M.; Agah, S.; Hosseini, A.F.; Hosseini, S.; Shidfar, S.; Aarbshahi, S.S.S.; Razmpoosh, E.; Shidfar, F. Effects of symbiotic and vitamin E supplementation on blood pressure, nitric oxide and inflammatory factors in non-alcoholic fatty liver disease. EXCLI J. 2017, 16, 278–290.
- Zhang, H.; Tsao, R. Dietary polyphenols, oxidative stress and antioxidant and anti-inflammatory effects. Curr. Opin. Food Sci. 2016, 8, 33–42.
- Wang, Z.; Zeng, M.; Wang, Z.; Qin, F.; Chen, J.; He, Z. Dietary Polyphenols to Combat Nonalcoholic Fatty Liver Disease via the Gut-Brain-Liver Axis: A Review of Possible Mechanisms. J. Agric. Food Chem. 2021, 69, 3585–3600.
- Abenavoli, L.; Larussa, T.; Corea, A.; Procopio, A.C.; Boccuto, L.; Dallio, M.; Federico, A.; Luzza, F. Dietary Polyphenols and Non-Alcoholic Fatty Liver Disease. Nutrients 2021, 13, 494.
- Houghton, D.; Stewart, C.J.; Day, C.P.; Trenell, M. Gut Microbiota and Lifestyle Interventions in NAFLD. Int. J. Mol. Sci. 2016, 17, 447.
- Evans, C.C.; LePard, K.J.; Kwak, J.W.; Stancukas, M.C.; Laskowski, S.; Dougherty, J.; Moulton, L.; Glawe, A.; Wang, Y.; Leone, V.; et al. Exercise prevents weight gain and alters the gut microbiota in a mouse model of high fat diet-induced obesity. PLoS ONE 2014, 9, e92193.
- Campbell, S.C.; Wisniewski, P.J.; Noji, M.; McGuinness, L.R.; Häggblom, M.M.; Lightfoot, S.A.; Joseph, L.B.; Kerkhof, L.J. The Effect of Diet and Exercise on Intestinal Integrity and Microbial Diversity in Mice. PLoS ONE 2016, 11, e0150502.
- Petriz, B.A.; Castro, A.P.; Almeida, J.A.; Gomes, C.P.C.; Fernandes, G.R.; Kruger, R.H.; Pereira, R.W.; Franco, O.L. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genomics 2014, 15, 511.
- Lambert, J.E.; Myslicki, J.P.; Bomhof, M.R.; Belke, D.D.; Shearer, J.; Reimer, R.A. Exercise training modifies gut microbiota in normal and diabetic mice. Appl. Physiol. Nutr. Metab. 2015, 40, 749–752.
- Carbajo-Pescador, S.; Porras, D.; Garcia-Mediavilla, M.V.; Martinez-Florez, S.; Juarez-Fernandez, M.; Cuevas, M.J.; Mauriz, J.L.; Gonzalez-Gallego, J.; Nistal, E.; Sanchez-Campos, S. Beneficial effects of exercise on gut microbiota functionality and barrier integrity, and gut-liver crosstalk in an in vivo model of early obesity and non-alcoholic fatty liver disease. Dis. Model. Mech. 2019, 12, dmm039206.
- Winn, N.C.; Liu, Y.; Rector, R.S.; Parks, E.J.; Ibdah, J.A.; Kanaley, J.A. Energy-matched moderate and high intensity exercise training improves nonalcoholic fatty liver disease risk independent of changes in body mass or abdominal adiposity—A randomized trial. Metabolism 2018, 78, 128–140.
- Yaribeygi, H.; Atkin, S.L.; Simental-Mendía, L.E.; Sahebkar, A. Molecular mechanisms by which aerobic exercise induces insulin sensitivity. J. Cell. Physiol. 2019, 234, 12385–12392.
- Houttu, V.; Boulund, U.; Grefhorst, A.; Soeters, M.R.; Pinto-Sietsma, S.J.; Nieuwdorp, M.; Holleboom, A.G. The role of the gut microbiome and exercise in non-alcoholic fatty liver disease. Therap. Adv. Gastroenterol. 2020, 13, 1756284820941745.
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576.
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219.
- McLoughlin, M.R.; Orlicky, D.J.; Prigge, J.R.; Krishna, P.; Talago, E.A.; Cavigli, I.R.; Eriksson, S.; Miller, C.G.; Kundert, J.A.; Sayin, V.I.; et al. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. Proc. Natl. Acad. Sci. USA. 2019, 116, 11408–11417.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38.
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110.
- Tao, J.; Krutsenko, Y.; Moghe, A.; Singh, S.; Poddar, M.; Bell, A.; Oertel, M.; Singhi, A.D.; Geller, D.; Chen, X.; et al. Nuclear factor erythroid 2-related factor 2 and β-Catenin Coactivation in Hepatocellular Cancer: Biological and Therapeutic Implications. Hepatology 2021, 74, 741–759.
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324.
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118.
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843.
- Wilson, G.K.; Tennant, D.A.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406.
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173.
- Huang, Y.; Zhang, Y.; Ge, L.; Lin, Y.; Kwok, H.F. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers 2018, 10, 82.
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057.
- Zhu, C.; Ho, Y.J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.S.; Lowe, S.W.; Pajvani, U.B. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J. Hepatol. 2021, 74, 613–626.
- Pervez, M.A.; Khan, D.A.; Slehria, A.U.R.; Ijaz, A. Delta-tocotrienol supplementation improves biochemical markers of hepatocellular injury and steatosis in patients with nonalcoholic fatty liver disease: A randomized, placebo-controlled trial. Complement. Ther. Med. 2020, 52, 102494.
- Zhang, S.; Yang, Y.; Liang, Z.; Duan, W.; Yang, J.; Yan, J.; Wang, N.; Feng, W.; Ding, M.; Nie, Y.; et al. Silybin-mediated inhibition of Notch signaling exerts antitumor activity in human hepatocellular carcinoma cells. PLoS ONE 2013, 8, e83699.
- Bootcov, M.R.; Bauskin, A.R.; Valenzuela, S.M.; Moore, A.G.; Bansal, M.; He, X.Y.; Zhang, H.P.; Donnellan, M.; Mahler, S.; Pryor, K.; et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 11514–11519.
- Tzikas, S.; Vassilikos, V.; Keller, T. GDF-15 as a risk stratification biomarker for cardiovascular disease. Int. J. Cardiol. 2019, 292, 246–247.
- Ago, T.; Sadoshima, J. GDF15, a cardioprotective TGF-beta superfamily protein. Circ. Res. 2006, 98, 294–297.
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219.
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419.
- Chung, H.K.; Kim, J.T.; Kim, H.W.; Kwon, M.; Kim, S.Y.; Shong, M.; Kim, K.S.; Yi, H.S. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci. Rep. 2017, 7, 17238.
- Lee, H.Y.; Nga, H.T.; Tian, J.; Yi, H.S. Mitochondrial Metabolic Signatures in Hepatocellular Carcinoma. Cells 2021, 10, 1901.
- Si, Y.; Liu, X.; Cheng, M.; Wang, M.; Gong, Q.; Yang, Y.; Wang, T.; Yang, W. Growth differentiation factor 15 is induced by hepatitis C virus infection and regulates hepatocellular carcinoma-related genes. PLoS ONE 2011, 6, e19967.
- Liu, X.; Chi, X.; Gong, Q.; Gao, L.; Niu, Y.; Chi, X.; Cheng, M.; Si, Y.; Wang, M.; Zhong, J.; et al. Association of serum level of growth differentiation factor 15 with liver cirrhosis and hepatocellular carcinoma. PLoS ONE 2015, 10, e0127518.
- Lee, E.S.; Kim, S.H.; Kim, H.J.; Kim, K.H.; Lee, B.S.; Ku, B.J. Growth Differentiation Factor 15 Predicts Chronic Liver Disease Severity. Gut Liver 2017, 11, 276–282.
- Koo, B.K.; Um, S.H.; Seo, D.S.; Joo, S.K.; Bae, J.M.; Park, J.H.; Chang, M.S.; Kim, J.H.; Lee, J.; Jeong, W., II; et al. Growth differentiation factor 15 predicts advanced fibrosis in biopsy-proven non-alcoholic fatty liver disease. Liver Int. 2018, 38, 695–705.
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448.
- Smirne, C.; Rigamonti, C.; De Benedittis, C.; Sainaghi, P.P.; Bellan, M.; Burlone, M.E.; Castello, L.M.; Avanzi, G.C. Gas6/TAM Signaling Components as Novel Biomarkers of Liver Fibrosis. Dis. Markers 2019, 2019, 2304931.
- Day, C.P. From fat to inflammation. Gastroenterology 2006, 130, 207–210.
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 2003, 163, 1301–1311.
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90.
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239.
- Couchie, D.; Lafdil, F.; Martin-Garcia, N.; Laperche, Y.; Zafrani, E.S.; Mavier, P. Expression and role of Gas6 protein and of its receptor Axl in hepatic regeneration from oval cells in the rat. Gastroenterology 2005, 129, 1633–1642.
- Lafdil, F.; Chobert, M.N.; Deveaux, V.; Zafrani, E.S.; Mavier, P.; Nakano, T.; Laperche, Y.; Brouillet, A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J. Hepatol. 2009, 51, 55–66.
- Fourcot, A.; Couchie, D.; Chobert, M.N.; Zafrani, E.S.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G1043–G1053.
- Gogoi-Tiwari, J.; Köhn-Gaone, J.; Giles, C.; Schmidt-Arras, D.; Gratte, F.D.; Elsegood, C.L.; McCaughan, G.W.; Ramm, G.A.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. The Murine Choline-Deficient, Ethionine-Supplemented (CDE) Diet Model of Chronic Liver Injury. J. Vis. Exp. 2017, 128, 56138.
- Soret, P.A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36.
- Eng, J.M.; Estall, J.L. Diet-Induced Models of Non-Alcoholic Fatty Liver Disease: Food for Thought on Sugar, Fat, and Cholesterol. Cells 2021, 10, 1805.
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076.
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579.
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424.
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; Van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522.
- Maquoi, E.; Vörös, G.; Carmeliet, P.; Collen, D.; Lijnen, H.R. Role of Gas-6 in adipogenesis and nutritionally induced adipose tissue development in mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1002–1007.
- Castrillo, A.; Tontonoz, P. Nuclear receptors in macrophage biology: At the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 2004, 20, 455–480.
- Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258.
- Zizzo, G.; Cohen, P.L. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: A key role for PPAR-γ in human macrophage polarization. J. Inflamm. 2015, 12, 36.
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.A.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240.
- Szanto, A.; Roszer, T. Nuclear receptors in macrophages: A link between metabolism and inflammation. FEBS Lett. 2008, 582, 106–116.
- Repa, J.J.; Mangelsdorf, D.J. The liver X receptor gene team: Potential new players in atherosclerosis. Nat. Med. 2002, 8, 1243–1248.
- Pastore, M.; Grimaudo, S.; Pipitone, R.M.; Lori, G.; Raggi, C.; Petta, S.; Marra, F. Role of Myeloid-Epithelial-Reproductive Tyrosine Kinase and Macrophage Polarization in the Progression of Atherosclerotic Lesions Associated With Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2019, 10, 604.
- Ait-Oufella, H.; Pouresmail, V.; Simon, T.; Blanc-Brude, O.; Kinugawa, K.; Merval, R.; Offenstadt, G.; Lesèche, G.; Cohen, P.L.; Tedgui, A.; et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1429–1431.
- Garbin, U.; Baggio, E.; Stranieri, C.; Pasini, A.; Manfro, S.; Mozzini, C.; Vallerio, P.; Lipari, G.; Merigo, F.; Guidi, G.; et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: A role for oxidized polyunsaturated fatty acids? Cardiovasc. Res. 2013, 97, 125–133.
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Yamada, M.; Mori, K.; Tanaka, S.; Maekawa, T.; Ebisutani, Y.; et al. Use of Mac-2 binding protein as a biomarker for nonalcoholic fatty liver disease diagnosis. Hepatol. Commun. 2017, 1, 780–791.
- Hayashi, S.; Nagaoka, K.; Tanaka, Y. Blood-Based Biomarkers in Hepatitis B Virus-Related Hepatocellular Carcinoma, Including the Viral Genome and Glycosylated Proteins. Int. J. Mol. Sci. 2021, 22, 11051.
- Bellan, M.; Castello, L.M.; Pirisi, M. Candidate Biomarkers of Liver Fibrosis: A Concise, Pathophysiology-oriented Review. J. Clin. Transl. Hepatol. 2018, 6, 317–325.
- Abe, M.; Miyake, T.; Kuno, A.; Imai, Y.; Sawai, Y.; Hino, K.; Hara, Y.; Hige, S.; Sakamoto, M.; Yamada, G.; et al. Association between Wisteria floribunda agglutinin-positive Mac-2 binding protein and the fibrosis stage of non-alcoholic fatty liver disease. J. Gastroenterol. 2015, 50, 776–784.
- Sugiura, T.; Dohi, Y.; Takase, H.; Yamashita, S.; Murai, S.; Tsuzuki, Y.; Ogawa, S.; Tanaka, Y.; Ohte, N. Serum levels of Mac-2 binding protein increase with cardiovascular risk and reflect silent atherosclerosis. Atherosclerosis 2016, 251, 192–196.
- Sugiura, T.; Dohi, Y.; Takase, H.; Yamashita, S.; Tsuzuki, Y.; Ogawa, S.; Tanaka, Y.; Ohte, N. Factors associated with longitudinal changes in serum concentrations of Mac-2 binding protein: A prospective 3-year observational study. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1337–1344.
- Arai, T.; Atsukawa, M.; Tsubota, A.; Kawano, T.; Koeda, M.; Yoshida, Y.; Tanabe, T.; Okubo, T.; Hayama, K.; Iwashita, A.; et al. Factors influencing subclinical atherosclerosis in patients with biopsy-proven nonalcoholic fatty liver disease. PLoS ONE 2019, 14, e0224184.
- Oh, S.; Tsujimoto, T.; Kim, B.; Uchida, F.; Suzuki, H.; Iizumi, S.; Isobe, T.; Sakae, T.; Tanaka, K.; Shoda, J. Weight-loss-independent benefits of exercise on liver steatosis and stiffness in Japanese men with NAFLD. JHEP Rep. Innov. Hepatol. 2021, 3, 100253.
- Ban, L.A.; Shackel, N.A.; McLennan, S.V. Extracellular Vesicles: A New Frontier in Biomarker Discovery for Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 376.
- Garcia-Martinez, I.; Alen, R.; Rada, P.; Valverde, A.M. Insights Into Extracellular Vesicles as Biomarker of NAFLD Pathogenesis. Front. Med. 2020, 7, 395.
- Masyuk, A.I.; Masyuk, T.V.; Larusso, N.F. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J. Hepatol. 2013, 59, 621–625.
- Szabo, G.; Momen-Heravi, F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455–466.
- Moratti, E.; Vezzalini, M.; Tomasello, L.; Giavarina, D.; Sorio, C. Identification of protein tyrosine phosphatase receptor gamma extracellular domain (sPTPRG) as a natural soluble protein in plasma. PLoS ONE 2015, 10, e0119110.
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles from Hepatocytes. Gastroenterology 2016, 150, 956–967.
- Ibrahim, S.H.; Hirsova, P.; Tomita, K.; Bronk, S.F.; Werneburg, N.W.; Harrison, S.A.; Goodfellow, V.S.; Malhi, H.; Gores, G.J. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016, 63, 731–744.
- Zhao, Y.; Zhao, M.F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.W.; Shen, D.; Zhang, J.Z.; Zhou, N.; He, J.; et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat. Commun. 2020, 11, 719.
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141.
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic. Res. 2019, 53, 841–850.
- Ore, A.; Akinloye, O.A. Oxidative Stress and Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina 2019, 55, 26.
- Korish, A.A.; Arafah, M.M. Camel milk ameliorates steatohepatitis, insulin resistance and lipid peroxidation in experimental non-alcoholic fatty liver disease. BMC Complement. Altern. Med. 2013, 13, 264.
- Schröder, T.; Kucharczyk, D.; Bär, F.; Pagel, R.; Derer, S.; Jendrek, S.T.; Sünderhauf, A.; Brethack, A.K.; Hirose, M.; Möller, S.; et al. Mitochondrial gene polymorphisms alter hepatic cellular energy metabolism and aggravate diet-induced non-alcoholic steatohepatitis. Mol. Metab. 2016, 5, 283–295.
- Elshazly, S.M. Ameliorative effect of nicorandil on high fat diet induced non-alcoholic fatty liver disease in rats. Eur. J. Pharmacol. 2015, 748, 123–132.
- Sugatani, J.; Wada, T.; Osabe, M.; Yamakawa, K.; Yoshinari, K.; Miwa, M. Dietary inulin alleviates hepatic steatosis and xenobiotics-induced liver injury in rats fed a high-fat and high-sucrose diet: Association with the suppression of hepatic cytochrome P450 and hepatocyte nuclear factor 4alpha expression. Drug Metab. Dispos. 2006, 34, 1677–1687.
- Li, W.; Lu, Y. Hepatoprotective Effects of Sophoricoside against Fructose-Induced Liver Injury via Regulating Lipid Metabolism, Oxidation, and Inflammation in Mice. J. Food Sci. 2018, 83, 552–558.
- García-Ruiz, I.; Rodríguez-Juan, C.; Díaz-Sanjuan, T.; Del Hoyo, P.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006, 44, 581–591.
- Verbeek, J.; Spincemaille, P.; Vanhorebeek, I.; Van Den Berghe, G.; Vander Elst, I.; Windmolders, P.; Van Pelt, J.; Van Der Merwe, S.; Bedossa, P.; Nevens, F.; et al. Dietary intervention, but not losartan, completely reverses non-alcoholic steatohepatitis in obese and insulin resistant mice. Lipids Health Dis. 2017, 16, 46.
- Ciapaite, J.; Bakker, S.J.L.; Van Eikenhorst, G.; Wagner, M.J.; Teerlink, T.; Schalkwijk, C.G.; Fodor, M.; Ouwens, D.M.; Diamant, M.; Heine, R.J.; et al. Functioning of oxidative phosphorylation in liver mitochondria of high-fat diet fed rats. Biochim. Biophys. Acta 2007, 1772, 307–316.
- Janevski, M.; Antonas, K.N.; Sullivan-Gunn, M.J.; McGlynn, M.A.; Lewandowski, P.A. The effect of cocoa supplementation on hepatic steatosis, reactive oxygen species and LFABP in a rat model of NASH. Comp. Hepatol. 2011, 10, 10.
- García-Ruiz, I.; Fernández-Moreira, D.; Solís-Muñoz, P.; Rodríguez-Juan, C.; Díaz-Sanjuán, T.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. Mitochondrial complex I subunits are decreased in murine nonalcoholic fatty liver disease: Implication of peroxynitrite. J. Proteome Res. 2010, 9, 2450–2459.
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193.
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509.
- Cardoso, A.R.; Kakimoto, P.A.H.B.; Kowaltowski, A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: Role of very long chain acyl-CoA dehydrogenases. PLoS ONE 2013, 8, e77088.
- Kathirvel, E.; Chen, P.; Morgan, K.; French, S.W.; Morgan, T.R. Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. J. Gastroenterol. Hepatol. 2010, 25, 1136–1143.
- Yoshioka, S.; Hamada, A.; Jobu, K.; Yokota, J.; Onogawa, M.; Kyotani, S.; Miyamura, M.; Saibara, T.; Onishi, S.; Nishioka, Y. Effects of Eriobotrya japonica seed extract on oxidative stress in rats with non-alcoholic steatohepatitis. J. Pharm. Pharmacol. 2010, 62, 241–246.
- Chung, M.Y.; Park, H.J.; Manautou, J.E.; Koo, S.I.; Bruno, R.S. Green tea extract protects against nonalcoholic steatohepatitis in ob/ob mice by decreasing oxidative and nitrative stress responses induced by proinflammatory enzymes. J. Nutr. Biochem. 2012, 23, 361–367.
- Song, L.; Qu, D.; Zhang, Q.; Jiang, J.; Zhou, H.; Jiang, R.; Li, Y.; Zhang, Y.; Yan, H. Phytosterol esters attenuate hepatic steatosis in rats with non-alcoholic fatty liver disease rats fed a high-fat diet. Sci. Rep. 2017, 7, 41604.
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; De Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum oxidative stress markers and lipidomic profile to detect NASH patients responsive to an antioxidant treatment: A pilot study. Oxid. Med. Cell. Longev. 2014, 2014, 169216.
- Leghi, G.E.; Domenici, F.A.; Vannucchi, H. Influence of oxidative stress and obesity in patients with nonalcoholic steatohepatitis. Arq. Gastroenterol. 2015, 52, 228–233.
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438.
- Loguercio, C.; De Girolamo, V.; De Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Pia Di Marino, M.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574.
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62.
- Oliveira, C.P.M.S.; Faintuch, J.; Rascovski, A.; Furuya, C.K.; Do Socorro Bastos, M.; Matsuda, M.; Della Nina, B.I.; Yahnosi, K.; Abdala, D.S.P.; Vezozzo, D.C.P.; et al. Lipid peroxidation in bariatric candidates with nonalcoholic fatty liver disease (NAFLD)—Preliminary findings. Obes. Surg. 2005, 15, 502–505.
- Dasarathy, S.; Yang, Y.; McCullough, A.J.; Marczewski, S.; Bennett, C.; Kalhan, S.C. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 382–388.
- Lu, Y.; Liu, X.; Jiao, Y.; Xiong, X.; Wang, E.; Wang, X.; Zhang, Z.; Zhang, H.; Pan, L.; Guan, Y.; et al. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARα. J. Clin. Investig. 2014, 124, 3501–3513.
- Zhu, J.Z.; Zhu, H.T.; Dai, Y.N.; Li, C.X.; Fang, Z.Y.; Zhao, D.J.; Wan, X.Y.; Wang, Y.M.; Wang, F.; Yu, C.H.; et al. Serum periostin is a potential biomarker for non-alcoholic fatty liver disease: A case-control study. Endocrine 2016, 51, 91–100.
- Yang, Z.; Zhang, H.; Niu, Y.; Zhang, W.; Zhu, L.; Li, X.; Lu, S.; Fan, J.; Li, X.; Ning, G.; et al. Circulating periostin in relation to insulin resistance and nonalcoholic fatty liver disease among overweight and obese subjects. Sci. Rep. 2016, 6, 37886.
- Luo, Y.; Qu, H.; Wang, H.; Wei, H.; Wu, J.; Duan, Y.; Liu, D.; Deng, H. Plasma Periostin Levels Are Increased in Chinese Subjects with Obesity and Type 2 Diabetes and Are Positively Correlated with Glucose and Lipid Parameters. Mediators Inflamm. 2016, 2016, 6423637.
- Dongiovanni, P.; Anstee, Q.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238.
- Salomone, F.; Li Volti, G.; Rosso, C.; Grosso, G.; Bugianesi, E. Unconjugated bilirubin, a potent endogenous antioxidant, is decreased in patients with non-alcoholic steatohepatitis and advanced fibrosis. J. Gastroenterol. Hepatol. 2013, 28, 1202–1208.
- Chtioui, H.; Semela, D.; Ledermann, M.; Zimmermann, A.; Dufour, J.F. Expression and activity of the cytochrome P450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007, 27, 764–771.
- Marcolin, É.; Forgiarini, L.F.; Tieppo, J.; Dias, A.S.; de Freitas, L.A.R.; Marroni, N.P. Methionine- and choline-deficient diet induces hepatic changes characteristic of non-alcoholic steatohepatitis. Arq. Gastroenterol. 2011, 48, 72–79.
- Nosrati, N.; Aghazadeh, S.; Yazdanparast, R. Effects of Teucrium polium on Insulin Resistance in Nonalcoholic Steatohepatitis. J. Acupunct. Meridian Stud. 2010, 3, 104–110.
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736.
- Marcolin, E.; San-Miguel, B.; Vallejo, D.; Tieppo, J.; Marroni, N.; González-Gallego, J.; Tuñón, M.J. Quercetin treatment ameliorates inflammation and fibrosis in mice with nonalcoholic steatohepatitis. J. Nutr. 2012, 142, 1821–1828.
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohli, K.K.; Bhansali, A. Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have Higher Oxidative Stress in Comparison to Chronic Viral Hepatitis. J. Clin. Exp. Hepatol. 2013, 3, 12–18.
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.K.M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62.
- Sumida, Y.; Nakashima, T.; Yoh, T.; Furutani, M.; Hirohama, A.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Okanoue, T.; et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J. Hepatol. 2003, 38, 32–38.
- Köroǧlu, E.; Canbakan, B.; Atay, K.; Hatemi, I.; Tuncer, M.; Dobrucali, A.; Sonsuz, A.; Gültepe, I.; Şentürk, H. Role of oxidative stress and insulin resistance in disease severity of non-alcoholic fatty liver disease. Turk. J. Gastroenterol. 2016, 27, 361–366.
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855.

