During liver injury, oxidative stress induces the activation of redox-sensitive transcription factors, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein-1 (AP-1), leading to an inflammatory response and the activation of cell death pathways in hepatocytes. In NAFLD, ROS regulates NF-kB activation by increasing the expression of pro-inflammatory cytokine TNF-α
[61]. NF-κB, a significant regulator of the inflammatory response, plays a vital role in regulating the transcription of genes involved in the establishment of the immune and inflammatory responses
[62]. Reduced NF-κB activity by antioxidants has been proposed as a therapeutic target in NASH due to its anti-inflammatory properties
[63]. Moreover, in the development of steatohepatitis, Nrf2 acts as a significant regulator of the redox balance and mediates anti-inflammatory and antiapoptotic effects of antioxidants
[64]. Upon oxidative stress, the cytoplasmatic transcription factor Nrf2, which regulates the expression of genes encoding antioxidant enzymes and regulators of the GSH (such as glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase modifier subunit, GPX7), translocates into the nucleus, where it interacts with specific DNA sequences called antioxidant response elements (ARE) in the promoter of its target antioxidant enzyme genes
[65]. Nrf2 expression is increased in the first stage of NAFLD in preclinical models, and pharmacological activation of Nrf2 in mice fed a high-fat and high-fructose diet decreases NASH parameters (insulin resistance, weight, triglycerides, ALT levels) via the transcriptional regulation of genes involved in inflammation, apoptosis, fibrosis, ER stress, and OS
[66]. Consistently,
Nrf2-knockout mice treated with MCD show exacerbation of liver inflammation and steatosis, compared to control mice
[67]. In addition,
Nrf2-deficient mice fed a HFD develop a more severe NASH phenotype than their wild-type counterparts
[68]. Other evidence has shown that the dysfunctional Nrf2 in patients with NASH is tightly involved in the grade of inflammation but not of steatosis
[69]. In addition, upregulated Nrf2 in senescent hepatocytes is related to the activation of cocultured HSC. The Nrf2 agonist sulforaphane remarkably inhibits the effect of lipid accumulation-induced hepatocyte senescence on activation of HSC by the Nrf2-ARE pathway
[70]. Nrf2 also directly affects lipid metabolism by the activation of genes involved in FA oxidation (acyl-CoA oxidase 2, carnitine palmitoyltransferase 1), triglyceride export (apolipoprotein B), and the lipogenic transcription factor sterol regulatory element binding transcription factor 1 (SREBP-1)
[66].
Nfr2 deficiency in HFD-fed mice diminishes phosphorylation of acetyl-CoA carboxylate (ACC), a rate-limiting enzyme of hepatic FA synthesis, and, thus, increases its activity
[68]. A further study reported that the dysfunction of redox homeostasis induces hepatocytes to be highly susceptible to proteasome-associated metabolic stress. In comparison, insufficient peroxisome proliferator activating ligand receptors (PPAR) γ/Nrf2-driven antioxidative response is the main factor
[71]. Moreover, the interaction between NF-κB and Nrf2 is also a noticeable target for NAFLD progression. Evidence showed that NF-κB p65 subunit represses the Nrf2/ARE system at the transcriptional level by competitive interaction with the binding domain of the CREB-binding protein (CBP)
[72]. NF-κB dissociates from inhibitor kappa B (IκB) and then translocates to the nucleus. Nrf2 negatively controls the NF-κB signaling pathway by multiple mechanisms, including inhibiting nuclear translocation of NF-κB and blocking the degradation of IκB-α
[73].
Collectively, these data indicate that alterations in antioxidant pathways are associated with NAFLD, suggesting a role of oxidative stress in disease progression.
2.4. Relationship of Oxidative Stress and the Development and/or Progression of NAFLD-HCC
Taking into account the aforementioned epidemiological data that clearly indicate that NAFLD/NASH is causing a dramatic increase in the prevalence of HCC development, with NASH being a recognized etiological cause of HCC, it is quite understandable why many studies have attempted to unravel the possible role of OS also in the development of HCC, similarly to what happened for NAFLD and NASH. While increased OS in liver parenchymal cells has been observed and linked to HCC, both the detailed mechanisms and the overall impact of this specific problem have yet to be fully clarified
[175]. It is well known that ROS, such as H202, can cause point mutations or larger lesions in the genome
[176]. A recent study investigated in detail the role of OS-related enzymes and receptors in liver carcinogenesis, identifying three factors (thioredoxin reductase-1, glutathione reductase, and the transcription factor Nrf2) as major players in the development of HCC
[177]. The authors concluded that this process is the result of a complex interaction between several factors. Indeed, although it is well known that anti-oxidant systems constitute an integrated and finely tuned network capable of effectively preventing carcinogenesis by protecting healthy cells, the role of OS is controversial in existing cancers, where ROS are definitely part of the tumor microenvironment (TME)
[178] and the antioxidant network probably plays both anti- and pro-cancer roles. In this context, ROS are active players in cancer development, exerting apparently contradictory effects, i.e., stimulation of tumorigenesis and cancer cell proliferation or induction of cell death. Focusing on NAFLD/NASH, an indirect confirmation of the tumorigenic role of OS is the adaptation of cancer cells to antioxidant insults, e.g., by increasing NADPH through the pentose phosphate pathway (PPP), which is also an emerging mechanism of drug resistance exploited by cancer cells
[179]. In this context, dissecting the role of NOX (such as NOX1, NOX2, and NOX4), which are both NADPH consumers and ROS generators, would be of great interest.
In any case, an exhaustive analysis of all ROS involved in the transition from NASH to HCC far exceeds the aims of this review. Briefly, the most well-established signaling cascade regulated by ROS is Nrf2. When active in neoplastic cells, Nrf2 signaling restricts ROS damage and favors cancer cell survival under chronic OS. It is believed that some cancer cells actually use Nrf2 signaling as an adaptive mechanism to promote tumor growth
[180]. Recently, it was proposed that Nrf2 activation due to the microenvironment could be one of two hits required for HCC, while a mutation in the gene encoding β-catenin, a major genetic aberration observed in a significant subset of NAFLD-associated HCC, provides the second hit
[181]. NF-κB is another important pathway in this context. It can have both pro- and antioxidant roles by affecting the expression of target enzymes involved in ROS scavenging and generation
[182]. Despite this role of NF-κB in maintaining antioxidant defenses and reducing liver damage, obese and NAFLD patients display increased pro-inflammatory cytokine levels, hepatic NF-κB activation, and risk for HCC development
[183]. ROS can also indirectly and directly affect the tumor suppressor p53 activity (with higher expressions causing liver inflammation and NAFLD progression)
[184] and hypoxia-inducible factors (HIF) (with a positive correlation with HCC development and resistance to chemotherapeutic agents)
[185]. In addition to classical ROS pathways, B-cell lymphoma 2 (BCL-2) proteins are emerging as physiological- and pathophysiological-associated redox molecules in cell survival and death, with a possible role in NAFLD/NASH tumorigenesis. As a matter of fact, BCL-2 proteins can contribute to NAFLD hepatic ROS formation; in addition, ROS levels can trigger cell death through BCL-2 protein modulation
[186]. Protein tyrosine phosphatase (PTP) oxidation is another key mechanism of downstream ROS production with a proposed role in the transition from SS to NASH and HCC
[187]. Briefly, OS stress inhibition of PTPs serves as a mechanism by which optimal tyrosine phosphorylation is maintained under physiological conditions but, if dysregulated, can contribute to liver dysfunction and HCC development, especially in obese subjects
[188].
Among other possible, interesting new molecular mechanisms linking OS and NAFLD tumorigenesis there is the Notch signaling pathway. Selective blocking of Notch homolog 1, translocation-associated (Notch1), inhibits cancer cell growth and deregulates angiogenesis
[189]. By performing RNA sequencing of HFD-fed mice, Zhu et al. demonstrated that Notch-active hepatocytes show transcriptional enrichment of ECM-related genes, which may represent a mechanism that persists in the tumorigenic process. Furthermore, the same animal model with Notch active mutation spontaneously forms fully developed liver tumors
[190]. Therefore, it can be inferred that the continuous activation of the Notch signaling pathway promotes the occurrence of NAFLD-related HCC. A further indirect evidence comes from the potential inhibition of HCC invasion and metastasis by δ-tocotrienol, an isomer of VitE, via downregulating the Notch1 signaling pathway with reduced biochemical markers of hepatocellular injury and steatosis
[191], and by silymarin via inhibiting factors such as Notch intra-cellular domain (NICD), recombining binding protein suppressor of hairless (RBP-Jκ) (i.e., the central player in the transcriptional regulation of Notch target genes and functions) and hairy and enhancer of split-1 (Hes1)
[192].
2.5. Biomarkers of Oxidative Stress in NAFLD/NASH
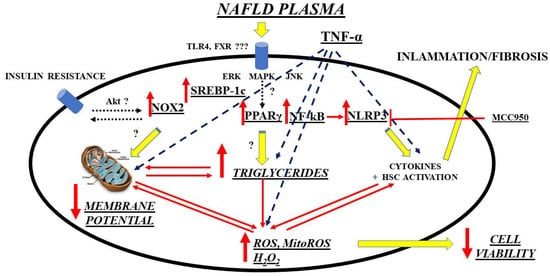
A further interesting and innovative line of research is the investigation of possible new biomarkers that have a diagnostic/prognostic role in relation to OS in NAFLD/NASH and that go alongside those already more validated regarding liver fibrosis. Though not well defined, circulating factors in NAFLD patients could modulate intracellular pathways in the direction predicted by current hypotheses on NAFLD pathogenesis, which has been recently evidenced by an in vitro study. Hence, exposure of primary human hepatocytes and Huh7.5 cells to plasma of NAFLD patients was able to cause triglycerides’ accumulation and OS through mechanisms involving SREBP-1c, inflammasome nucleotide-binding oligomerization domain-containing protein (NOD), leucine-rich repeats (LRR), pyrin domain-containing protein 3 (NLRP3), and PPAR γ, as a starting point
[95]. (
Figure 1).
Figure 1. Mechanisms of damage elicited by NAFLD plasma on hepatocytes. ERK: extracellular signal-regulated kinases; FXR: farnesoid X receptor; HSC: hepatic stellate cells; MAPK: mitogen-activated protein kinase; ROS: reactive oxygen species; MitoROS: mitochondrial ROS; H2O2: hydrogen peroxide; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: NOD-LRR-and pyrin domain-containing protein 3; MCC950: a specific, small-molecule inhibitor of NLRP3 inflammasome; NOX: nicotinamide adenine dinucleotide phosphate (NADPH) oxidases; Akt: Ak strain transforming factor; PPAR: peroxisome proliferator activating ligand receptors; SREBP: sterol regulatory element binding protein; TLR: toll-like receptor; TNF: tumor necrosis factor; JNK: c-Jun N-terminal kinase.
Following these premises, a promising, novel, systemic marker of OS, but also of inflammation and cellular aging, is represented by growth differentiation factor-15 (GDF-15)
[193][194][195]. This is a cytokine upregulated in multiple pathological conditions where OS, endothelial dysfunction, tissue aging, and chronic inflammation are the hallmarks. As a matter of fact, GDF-15 is rapidly produced by various cell types, including hepatocytes, cardiomyocytes, macrophages, adipocytes, and endothelial and smooth cells, among others, in response to pro-inflammatory cytokines or ROS, cellular stress, tissue injury, hypoxia, and/or oncogene activation
[196]. More in detail, this factor has been identified as a mitochondrial unfolded protein response (UPR
mt)-associated cell non-autonomous mitokine that regulates systemic energy homeostasis and feeding behavior
[197]. Due to the link between mitochondrial dysfunction and liver injury, GDF15 is currently considered a biomarker of diverse chronic liver diseases
[198]. First of all, this factor has been demonstrated, as expected, to be a possible prognostic HCC marker because its upregulation as a consequence of mitochondrial oxidative phosphorylation defects and ROS production is associated with liver cancer development, progression, and metastasis
[199]. Taking into consideration the same physiopathological considerations, it is not surprising that the factor has also a potential prognostic role in hepatitis B and C chronic viral hepatitis, with an increasing degree of diagnostic accuracy in compensated and decompensated liver cirrhosis
[200][201][202]. Finally, its pathogenic role was recently demonstrated also in the development and progression of NAFLD. With concern to the latter condition, GDF15 increased the risk of NASH and advanced fibrosis among biopsy-proven NAFLD patients, independently of known metabolic risk factors [(such as age, gender, body mass index (BMI), insulin resistance, and low skeletal muscle mass)]; moreover, the factor was associated with the severity of lobular inflammation and ballooning
[203][204].
Among other promising biomarkers of OS in NAFLD, the growth arrest-specific gene 6 (Gas6) serum protein and its family of tyrosine kinase receptors, namely, Tyro3, Axl, and MERTK (TAM), can be cited. The Gas6/TAM system (mainly, Axl and MERTK) is already a well-known important player in the progression of liver fibrosis in various hepatic diseases, including NAFLD/NASH
[205]. Focusing on OS, it is necessary to go back for a moment to the aforementioned “two hits” theory of NASH. Briefly, steatosis is the first hit that generally is recognized to increase hepatocyte vulnerability to any secondary insult, eliciting an inflammatory response, characterized by lobular inflammation, elevated local and systemic cytokines, activation of HSC, and expansion of liver progenitor cells (LPC). Most probably, both events are strictly interconnected since fat accumulation per se induces OS, inflammatory cytokine synthesis, and LPC expansion
[206]. Well, the Gas6/TAM system could be a key element in this interplay
[207][208]. What is already known is that in the liver, Gas6 and at least its high-affinity receptor Axl, are expressed by many of the previously cited inflammatory elements, such as macrophages, HSC in their myofibroblastic phenotype
[209], and LPC
[210]. Moreover, in murine models, Gas6 deficiency reduced inflammation and activation of Kupffer cells and myofibroblasts, causing, among other things, delayed liver repair in response to acute injury
[211]. Based on these preliminary studies, which suggested a prominent role of Gas6 in the pathogenesis of chronic liver diseases including NAFLD/NASH, new studies followed, still analyzing the possible roles of Gas6 deficiency but this time focusing more on the possible changes in the oxidative state at the hepatic level. For instance, Gas6-deficient mice fed a choline deficient, ethionine-supplemented diet (CDE) showed not only, as expected, reduced liver inflammation and fibrosis, but also attenuated hepatic steatosis. More in detail, Gas6
−/− mice fed with the CDE diet showed a reduced downregulation of some crucial rate-limiting enzymes of mitochondrial FA β-oxidation, such as PPARα and its target genes acyl-CoA oxidase-1 (
ACOX1) and carnitine palmitoyltransferase-1 (
CPT1). Moreover, Gas6 deficiency reversed the CDE-induced repression of stearoyl-CoA desaturase-1 (
SCD1), the rate-limiting step in the biosynthesis of monounsaturated FA. Moreover, Gas6 deficiency at least partially mitigated the increased expression of CD36 hepatic fatty acid translocase (
CD36), an enzyme that mediates uptake and intracellular transport of long-chain FA in hepatocytes and that is known to be abnormally increased in NAFLD. All these mechanisms ultimately accounted for the more efficient FA catabolism and delayed steatosis found in Gas6 knockout mice
[212]. On the other hand, CDE diet is long known to promote intrahepatic lipid accumulation because of increased FA uptake and decreased triglycerides synthesis, in addition to the already mentioned inhibition of FA oxidation, and so can be a useful model if NAFLD-associated processes are the study focus
[213][214][215]. This happens probably because the CDE diet has been demonstrated to induce CD36 expression, favoring FA uptake, and to suppress the SCD1 mRNA, preventing synthesis of unsaturated FA
[216][217][218]. Concerning the latter point, a possible explanation for the higher SCD1 expression observed in Gas6
−/− mice is that they have a decreased expression of Kupffer cells-derived TNF-α, which, in turn, has been demonstrated to downregulate SCD1
[216]. Moreover, Gas6-deficient mice showed also reduced IL-1β expression, again produced from Kupffer cells. This a cytokine that promotes triglycerides’ storage in hepatocytes by reducing the expression of PPARα and its target genes involved in hepatocyte FA oxidation
[219]. Therefore, it can be assumed that reduced IL-1β expression revealed in Gas
−/− mice is an additional mechanism that prevents early CDE-induced downregulation of genes involved in β-oxidation in hepatocytes and steatosis. Of interest, a role of Gas6 in fat accumulation has been reported also in adipogenesis. After exposure to a HFD, fat accumulation in adipose tissues was reduced in Gas6 knockout mice, with no effect on hepatic steatosis. In this study, Gas6 directly promoted the proliferation of Axl-positive preadipocytes and their differentiation into mature adipocytes
[220]. For what concerns the liver, it was previously demonstrated that Gas6 and its receptor Axl are not expressed in hepatocytes. Therefore, the contribution of Gas6 to CDE-induced FA accumulation in these cells may not be direct but mediated by Axl-positive cells, such as macrophages, LPC, and HSC in their myofibroblastic phenotype
[209][210]. By the way, the total content of Axl was higher than in wild-type mice
[212]. New insights to the importance of the Gas6/TAM system pathway in the progression of NASH came also from studies involving the Gas6 receptor MERTK. In the metabolic syndrome, and more specifically in NAFLD, accumulation of excess lipoprotein-derived cholesterol in macrophages was shown to activate nuclear liver X receptors (LXR) that, in turn, triggered the induction of the ATP-binding cassette (ABC) transporter, mediating cholesterol efflux
[221] and the upregulation of MerTK both in mice
[222] and in humans
[223]. Both LXR and the aforementioned PPAR influence, among other things, the transcription of genes regulating lipid homeostasis and inflammation. More in detail, PPARγ can be activated by metabolic signals (i.e., polyunsaturated FA and lipoproteins)
[224], resulting in lipid uptake through the scavenger receptor CD36 and β-oxidation
[225]. LXRs are, in turn, oxysterol-activated transcription factors that sense elevated cellular cholesterol
[226]. PPARγ and LXR activities are generally coordinated and control the transcription of many receptors, including MerTK. In MERTK and LXRs, double knockout mice amplified pro-inflammatory responses, and increased susceptibility to OS and atherosclerosis were observed, suggesting that the LXR-dependent regulation of MerTK is important for normal homeostasis including FA metabolism
[227][228][229]. However, further studies are needed to confirm these preliminary data.
Mac-2 binding protein (M2BP), a known important player in cell adhesion, is another NAFLD biomarker candidate
[230]. For what concerns liver, a source of M2BP is HSC; since Kupffer cells have an increased M2BP production after autocrine or paracrine stimulation, it was suggested that M2BP secreted from HSC may be taken up by Kupffer cells
[231]. Based on these premises, the factor is mainly known as a liver fibrosis marker through inflammation in the extracellular matrix
[230][232][233]. However, there is now increasing evidence for a promising role also, as in the metabolic syndrome context. For instance, serum M2BP levels were higher in subjects with hypertension, dyslipidemia, or abnormal glucose metabolism compared to subjects without such risk factors. Moreover, the protein levels were associated with severity of cardiovascular risk. Subdivision of M2BP levels into quartiles revealed that M2BP was significantly associated with OS and, in particular, with reactive oxygen metabolites
[234]. Other studies confirmed that, in normal subjects, M2BP concentrations were not only associated with OS marker derivatives of reactive oxygen metabolites, but also with LDL-cholesterol and triglyceride levels. Furthermore, increased LDL-cholesterol was an independent determinant of M2BP high concentrations and, vice versa, increase in LDL-cholesterol was significantly greater in subjects in whom M2BP concentrations increased during the follow-up period
[235]. When focusing on NAFLD patients, there was an indirect association between M2BP levels and subclinical atherosclerosis as determined by brachial-ankle pulse wave velocity
[236]. A direct link between OS and M2BP was instead provided in a cohort of Japanese obese men with NAFLD. Independent of the effect of weight loss, a 3-month exercise regimen significantly reduced liver steatosis (by 9.5%) as determined by transient elastography. Moreover, an important decrease in OS parameters during the intervention was observed: Among other parameters, there was also serum M2BP (until −62.4% in the exercise group and −37.7% in the weight-loss group). These reductions paralleled with less significant decreases in insulin resistance (HOMA-IR) and lipid profile (triglycerides and non-esterified FA). Taken together, these data suggest that, in NAFLD, M2BP may be more a marker of organokine balance and systemic inflammation and OS than a surrogate liver steatosis parameter
[237] and could find a clinical application primarily in NAFLD patients with obesity and/or metabolic syndrome.
A new line of research in NAFLD biomarkers concerns extracellular vesicles (EVs). Several mechanisms implicated in NAFLD progression, such as inflammation, fibrosis, and angiogenesis, but also OS, all related to metabolic syndrome-associated lipotoxicity, trigger EV production and release by the liver
[238]. On the one hand, EVs mediate local intercellular communications between the liver cells, thereby driving disease pathogenesis and, on the other, liver-derived EVs could affect distant tissues and organs upon their release to the bloodstream. Thus, liver-derived EVs have been suggested as biomarkers both for diagnostic (potentially including early stages) and prognostic purposes in NAFLD patients (the so-called liquid biopsy)
[239][240]. However, the identification of liver-derived EVs in circulation as indicative of metabolic alterations in this organ is still a challenge for basic and clinical researchers. As previously cited, NAFLD is not an isolated condition and generally occurs as a complication of other metabolic disorders. Therefore, multiple tissues may be affected and, consequently, the contribution of extrahepatic EVs during NAFLD cannot be excluded (i.e., adipocyte- or immune cell-derived EVs). Furthermore, most liver cell types produce EVs including hepatocytes, cholangiocytes, HSC, and liver sinusoidal endothelial cells
[241]. Nonetheless, as 80% of the liver volume is composed by hepatocytes, their participation to the total pool of liver-derived EVs is likely the most relevant. Based on these premises and focusing on EVs of hepatocyte production, several protein-based EV biomarkers have been introduced for NAFLD liver damage
[242] or NASH
[243][244] with particular regard to the lipotoxicity link with inflammation, although, to date, most studies have focused on characterizing EVs-associated nucleic acids, especially microRNAs (miRNAs). For what concerns OS, some preliminary data exist precisely for EVs-derived miRNAs. It was demonstrated that liver firstly responds to lipid overload and thereafter sends hepatocyte-derived EVs (in particular, let-7e-5p), targeting adipocytes to regulate adipogenesis and lipogenesis. More in detail, these EVs positively correlated with BMI and enhanced adipocyte lipid deposition by increasing lipogenesis and inhibiting lipid oxidation through an axis involving also the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (
Pgc1α). Moreover, taking into account that lipid overload enhances liver geranylgeranyl diphosphate synthase (
Ggpps) expression, which, in turn, regulates EVs’ secretion through Rab27A geranylgeranylation, liver-specific
Ggpps-deficient mice had reduced fat adipose deposition because of reduced EVs’ secretion
[245]. Thus, this pilot study highlighted an inter-organ mechanism whereby the liver during NAFLD senses different metabolic states and sends corresponding signals to remodel adipose tissue to adapt to metabolic changes in response to lipid overload. In any case, future studies aiming to examine additional molecular mechanisms possibly involved in EVs’ biogenesis, release, and dysregulation of target cells as well as to identify cargos with potential value as biomarkers for noninvasive diagnosis and monitoring of NAFLD progression are much desirable.
There are many other biomarkers of OS that have been evaluated, or that are under evaluation, in animal and clinical models of NAFLD/NASH
[49][246][247][248]. These factors were detected primarily in the liver, plasma, and serum, although a few studies analyzed whole blood samples. Major approaches to measure these biomarkers included ELISA, colorimetry, and immunohistochemistry. Biomarkers of OS measured in experimental models of NAFLD/NASH included lipid oxidation products [thiobarbituric acid reactive substances (TBARS), which were used as MDA, 4-HNE, and 8-isoprostanes], DNA damage products [8-hydroxy-2′-deoxyguanosine (8-OH-dG)], and protein damage products (protein carbonyl, dityrosine, hydroxyproline, and nitrotyrosine) (
Table 1). Among these biomarkers, MDA was the most measured marker of OS. The level or activity of these factors was generally increased in most animal models, with a few exceptions
[249][250].
Table 1. Oxidative stress markers in animal models of NAFLD/NASH.
| Oxidative Stress Markers |
Disease Status |
Experimental
Model and Species |
Sample |
Changes in
Concentration/
Activity/Expression |
Reference(s) |
| TBARS/MDA |
Steatosis, NASH |
HFD, HF-HSD, MCD, ob/ob mice, CDHF diet |
Liver |
↑ |
[251][252] |
| HCD |
↓ |
| HFD, MCD |
NS |
| 4-HNE |
Steatosis, NASH |
HFD, MCD, HF-HSD, CDHF diet |
Liver |
↑ |
[253][254] |
| 8-OH-dG |
NASH |
MCD, ob/ob mice HFMCD |
Liver |
↑
↓ |
[255] |
| 8-isoprostane |
NASH |
HFMCD |
Liver |
↑ |
[256] |
| Protein carbonyl |
Steatosis, NASH |
HFD, MCD
HFD |
Liver |
↑
NS |
[257] |
| Nitrotyrosine |
NASH |
HFD, ob/ob mice |
Liver |
↑ |
[258] |
| Periostin |
NASH |
HFD, ob/ob mice |
Serum, liver |
↑ |
[259] |
| CYP2E1 |
Steatosis, NASH |
HFD, HF-HSD, CDHF |
Liver |
↑ |
[252][260] |
| Dityrosine |
Steatosis |
HFD |
Liver |
↑ |
[261] |
| Hydroxyproline |
NASH |
MCD/WD |
Liver |
↑ |
[262] |
| H2O2 |
NASH |
MCD |
Liver |
↑ |
[250] |
| Lipid peroxide |
NASH |
MCD |
Liver |
↑ |
[263] |
| NADPH oxidase |
NASH |
ob/ob mice |
Liver |
↑ |
[264] |
| Xanthine oxidase |
Steatosis |
HFD |
Liver |
↑ |
[265] |
For what concerns biomarkers of OS frequently determined in NAFLD/NASH patients, the most studied ones were quite similar to those previously mentioned for experimental models (
Table 2). Overall, the concentrations or activities of these biomarkers were increased in all data examined, although the rise was not significant in some cases
[53][266][267][268].
Table 2. Oxidative stress markers in NAFLD/NASH patients.
| Oxidative Stress Markers |
Disease Status |
Sample |
Changes in Concentration/
Activity/Expression |
Reference(s) |
| TBARS/MDA |
Steatosis, NASH |
Serum, liver, blood |
↑ |
[269][53][270] |
| Serum |
NS |
| 4-HNE |
NASH |
Liver |
↑ |
[271] |
| Hydroperoxides |
NASH |
Liver |
↑ |
[272] |
| 8-OH-dG |
Steatosis, NASH |
Liver, plasma
Liver |
↑
NS |
[273] |
| 8-isoprostane |
NASH |
Plasma |
NS |
[267] |
| Protein carbonyl |
Steatosis, NASH |
Liver |
↑ |
[268] |
| Nitrotyrosine |
Steatosis, NASH |
Liver |
↑ |
[25] |
| Blood |
NS |
| Periostin |
Steatosis, NASH |
Serum, plasma, liver
Serum, plasma |
↑ |
[274][275][276][277] |
| NS |
| Nitric oxide |
Steatosis, NASH |
Serum, blood |
↑ |
[278][279] |
| CYP2E1 |
Steatosis, NASH |
Liver |
↑ |
[280] |
| NS |
In addition, many studies, both in human and experimental models of NAFLD/NASH, also measured enzymatic and nonenzymatic liver antioxidant biomarkers [e.g., catalase, SOD, GSH peroxidase (GPx), GSH, thioredoxin reductase (TrxR), α-tocopherol, and ubiquinone], and their activities were decreased in most liver samples, consistently with a loss of their protective abilities due to high OS, while blood, serum, and plasma had more conflicting results
[257][265][267][281][282][283][284][285][286][287] (
Table 3 and
Table 4).
Table 3. Antioxidant markers in animal models of NAFLD/NASH.
| Antioxidant Marker |
Disease Status |
Experimental
Model and Species |
Sample |
Changes in
Concentration/
Activity/Expression |
Reference(s) |
| SOD |
Steatosis, NASH |
HF, HFD, OLETF rats, MCD |
Liver |
↓ |
[286][288] |
| HFD, MCD |
↑ |
| HFD |
NS |
| Catalase |
Steatosis, NASH |
HFD, MCD, HCD |
Liver |
↓ |
[53][285] |
| MCD |
↑ |
| GPx |
Steatosis, NASH |
HFD, MCD, HF |
Liver |
↓ |
[267][268] |
| MCD |
↑ |
| HFD |
NS |
| GSH |
Steatosis, NASH |
HFD, HCD, MCD, ob/ob mice |
Liver |
↓ |
[288] |
| OLETF rats, HF MCD |
↑ |
| GR |
NASH |
MCD |
Liver |
↓ |
[282] |
Table 4. Antioxidant markers in NAFLD/NASH patients.
| Antioxidant Marker |
Disease Status |
Sample |
Changes in
Concentration/
Activity/Expression |
Reference(s) |
| SOD |
Steatosis, NASH |
Serum, plasma, liver |
↓ |
[286][288] |
| Blood, serum |
↑ |
| Serum |
NS |
| Catalase |
Steatosis, NASH |
Plasma, blood, liver |
↓ |
[53][285] |
| Serum |
NS |
| GPx |
Steatosis, NASH |
Liver, serum |
↓ |
[267][268] |
| Blood |
↑ |
| Serum |
NS |
| GSH |
Steatosis, NASH |
Liver, blood |
↓ |
[288] |
| Serum |
↑ |
| GR |
Steatosis, NASH |
Serum, blood |
↑ |
[286] |
| TRX |
Steatosis |
Serum |
↑ |
[287] |
| α-Tocopherol |
Steatosis, NASH |
Serum |
↓ |
[275] |
| NS |
| Ubiquinone |
Steatosis |
Serum |
↓ |
[289] |
| Bilirubin |
Steatosis, NASH |
Serum |
↓ |
[279] |
| Ascorbic acid |
Steatosis, NASH |
Serum |
↓ |
[288] |
| NS |
Data derived from the measurement of all these markers clearly indicate that the presence of OS is strongly associated with NAFLD/NASH. However, these biomarkers provide very limited information on the type, amount, and localization of ROS as well as their targets and involvement in specific pathophysiological processes. Indeed, the cause-and-effect relationship between OS and pathogenesis has not yet been established with certainty, although many studies have provided possible mechanisms supporting the crucial role of OS in the pathogenesis of NAFLD.
Data derived from the measurement of all these markers clearly indicate that the presence of OS is strongly associated with NAFLD/NASH. However, these biomarkers provide very limited information on the type, amount, and localization of ROS as well as their targets and involvement in specific pathophysiological processes. Indeed, the cause-and-effect relationship between OS and pathogenesis has not yet been established with certainty, although many studies have provided possible mechanisms supporting the crucial role of OS in the pathogenesis of NAFLD.
3. Conclusions
Although the knowledge of NAFLD pathogenesis and natural history has greatly expanded in the last decade, many issues remain still unsolved. The purpose of this article was to summarize recent developments in the understanding of NAFLD, essentially focusing on OS as a major pathogenetic mechanism. Among different ROS generators, mitochondria could play a central role and contribute to liver injury caused by FA and/or a wide variety of their biologically active metabolites. Additionally, gut microbiota would be involved in inflammation and oxidation.
Starting from the assumption that OS would represent a trigger factor for NAFLD onset and evolution towards NASH and HCC, it is quite evident that the cornerstone of NAFLD treatment can be represented by the modulation of the balance between the antioxidants/oxidants, which is under the influence of individual genetic and epigenetic factors, as well. Various attempts to translate ROS scavenging by antioxidants into experimental and clinical studies have yielded mostly encouraging results. Among several antioxidants, the use of VitE has been particularly examined in Literature, although results obtained are somehow contradictory. Additionally, studies performed with natural polyphenols, which could be useful for NAFLD prevention and treatment thanks to their antioxidant activities, are quite satisfactory. In addition, probiotics/prebiotics, healthy diet, exercise, or fecal microbiota transplantation would represent new therapeutic approaches to modulate microbiota quality and diversity in order to prevent and/or avoid gut damage and consequent liver OS. Nevertheless, it must be said that most of the molecular targets and the exact cellular mechanisms of these compounds remain to be fully elucidated, and additional studies on their therapeutic potential are needed. Finally, given the importance of genetics in the NAFLD onset, precision medicine taking into consideration genetic/epigenetic factors will likely assist in targeting individualized, appropriate treatments.
As a final remark, it should be also highlighted that the finding of the role of any circulating factors in the NAFLD pathogenesis and evolution could open new frontiers for the early diagnosis of the disease and for the prognosis of patients. In this context, circulating EVs could represent the ideal candidate and would deserve further study.
In conclusion, at the current level of knowledge, the most important problem for OS in NAFLD remains the interpretation and correlation of some results obtained through experimental and clinical studies. A lot of these are difficult to translate into routine clinical practice, aimed at restoring a “healthy” lipid profile and/or reinforcing the antioxidant status, because of a loss of strong evidences that support their application in the therapy of this disease. Certainly, we must admit that the evolution in the comprehension of the mechanisms that support NAFLD has been very fast in the last decade, and, at the same time, the analyzed fields represent some of the most promising topics of scientific research of the future.