 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Waad Hussein Abuwatfa | + 5599 word(s) | 5599 | 2022-01-24 07:36:50 | | | |
| 2 | Rita Xu | Meta information modification | 5599 | 2022-01-24 10:21:58 | | |
Video Upload Options
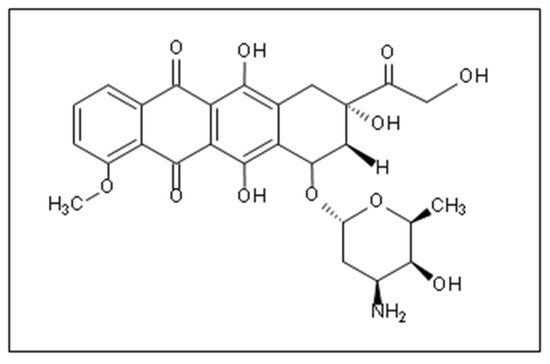
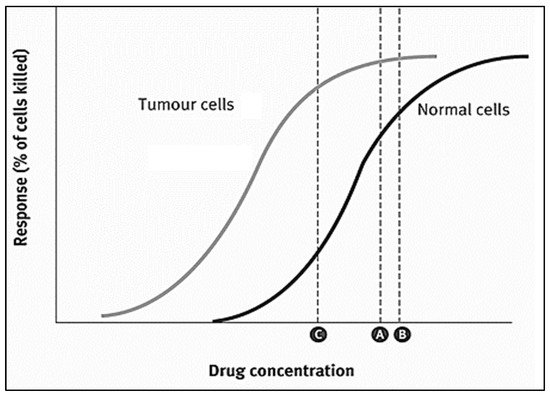
Doxorubicin (DOX) is one of the most widely used anthracycline anticancer drugs due to its high efficacy and evident antitumoral activity on several cancer types. However, its effective utilization is hindered by the adverse side effects associated with its administration, the detriment to the patients’ quality of life, and general toxicity to healthy fast-dividing cells. Thus, delivering DOX to the tumor site encapsulated inside nanocarrier-based systems (like liposomes, micelles, metal-organic frameworks (MOFs)) is an area of research that has garnered colossal interest in targeted medicine. Nanoparticles can be used as vehicles for the localized delivery and release of DOX, decreasing the effects on neighboring healthy cells and providing more control over the drug’s release and distribution.
1. Introduction
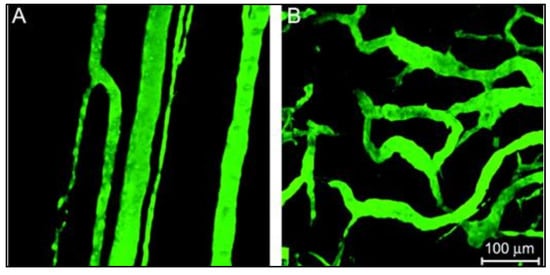
2. Nanoparticles as Drug Delivery Systems (DDS)
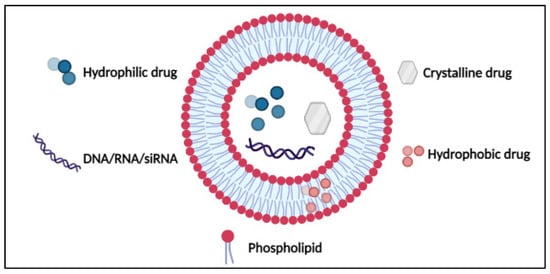
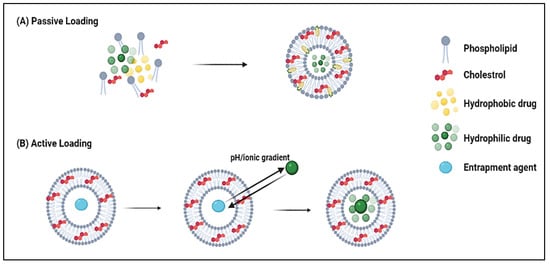
3. DOX Delivery Systems Based on Liposomes
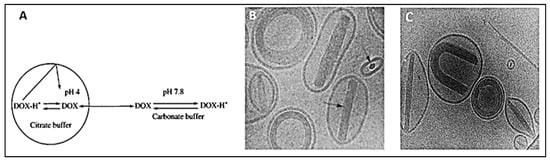
| Salt Gradient | Size ± SD (nm) | EE (%) |
|---|---|---|
| Ammonium Phosphate | 129.3 ± 3.7 | 98 |
| Ammonium Sulfate | 129.2 ± 2.9 | 95 |
| Ammonium Acetate | 115.9 ± 1.0 | 77 |
| Ammonium Citrate | 114.9 ± 1.2 | 100 |
| Sodium Phosphate | 113.4 ± 1.6 | 52 |
| Sodium Sulfate | 111.8 ± 1.9 | 44 |
| Sodium Acetate | 113.4 ± 1.6 | 16 |
| Sodium Citrate | 151.7 ± 3.8 | 54 |
| Preparation Method | Target Cancer | Functionalization | Study Model | Triggering Modality | Findings | Ref. |
|---|---|---|---|---|---|---|
| Ethanol injection | osteosarcoma | Estrogen | In vitro flow cytometry and MTT analysis on MG63 (estrogen overexpressing) cells and LO2 (negative liver cells). Ex vivo imaging of MG63 tumors extracted from Male BALB/c nude mice. |
Redox-sensitivity and glutathione responsiveness | Loaded decorated liposomes size~110 nm. Exhibited high encapsulation efficiency. Ex vivo analysis of the functionalized liposomes showed more selective accumulation in tumor tissues compared to other vital organs, and in vitro results showed higher cytotoxicity towards overexpressing cells. |
[66] |
| Thin-film hydration | Lymphoma | anti-CD19 moiety; PEG grafted by disulfide links (mPEG-S-S-DSPE) | In vitro MTT assay In vivo model: Female BALB/c Cr Alt B/M mice bearing Namalwa cells |
pH sensitivity | Liposomes decorated with cleavable PEG chains rapidly dissociated in the plasma. The pH-sensitive liposomes, targeting the CD19 epitope excessively abundant on B-lymphoma cells, showed increased selective cytotoxicity towards these cells, and enhanced release kinetics at lower pH levels. |
[67] |
| Post-insertion; mixing with preformed DOXIL | Cancer Stem Cells (CSCs) | anti-CD44 monoclonal antibody (mAb) | In vitro flow cytometry and MTT assay on C-26 and NIH-3T3 (non-tumor) cells. In vivo model: female BALB/c mice bearing C-26 colon carcinoma. |
N/A | Functionalization of DOXIL liposomes significantly increased their size. The IC50 values were lower on the C-26 cell line overexpressing CD44, while higher values were reported for the negative cell line (NIH-3T3). | [68] |
| Solvent evaporation | Various cancers | Cationic Polymethacrylate Eudragit RL100 | In vitro flow cytometry and MTT assay on MCF7/adr and H22 cells. In vivo model: ICR mice bearing aggressive liver cancer H22 cells. |
N/A | Functionalization of liposomes with Polymethacrylate derivatives increases their cellular internalization and antitumoral activity. The in vivo results showed that four injections of the functionalized formulation led to tumor size reduction by 60%. | [69] |
| Thin-film hydration | Metastatic lung cancer | CXCR4-antagonist cyclic peptide (peptide R) | In vitro cytotoxicity assay. In vivo model: C57BL/6 mice bearing B16 human melanoma cells |
N/A | In vitro results showed that targeting significantly decreased the IC50 while reducing metastasis and regression in tumor size growth. | [70] |
| Film dispersion | hepatocellular carcinoma (HCC) | glycyrrhetinic acid (GA) and peanut agglutinin (PNA) | In vitro specific uptake of HepG2, MCF-7, and SMMC-7721 cells In vivo model: male BALB/C-nu mice bearing SMMC-7721 xenografts. |
N/A | HepG2 cells showed the highest uptake towards the liposomes functionalized with GA alone, while MCF-7 showed the highest affinity towards the PNA functionalized liposomes. The dual-targeted liposomal formulation was most internalized by the SMMC-7721 | [71] |
References
- Husseini, G.A.; Jabbar, N.A.; Mjalli, F.S.; Pitt, W.G. Modeling and sensitivity analysis of acoustic release of Doxorubicin from unstabilized pluronic P105 using an artificial neural network model. Technol. Cancer Res. Treat. 2007, 6, 49–56.
- Hagos, E.G.; Ghaleb, A.M.; Dalton, W.B.; Bialkowska, A.B.; Yang, V.W. Mouse embryonic fibroblasts null for the Krüppel-like factor 4 gene are genetically unstable. Oncogene 2009, 28, 1197–1205.
- Sudhakar, A. History of Cancer, Ancient and Modern Treatment Methods. J. Cancer Sci. Ther. 2009, 1, 1–4.
- Espinosa, E.; Zamora, P.; Feliu, J.; Barón, M.G. Classification of anticancer drugs—a new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523.
- Espinosa, E.; Raposo, C.G. Classification of Anticancer Drugs Based on Therapeutic Targets. In Macromolecular Anticancer Therapeutics; Springer: New York, NY, USA, 2009; pp. 3–35.
- Wu, X.-Z. A new classification system of anticancer drugs—Based on cell biological mechanisms. Med. Hypotheses 2006, 66, 883–887.
- Bhattacharya, B.; Mukherjee, S. Cancer Therapy Using Antibiotics. J. Cancer Ther. 2015, 6, 849–858.
- Mier, W.; Hoffend, J.; Haberkorn, U.; Eisenhut, M. Current Strategies in Tumor-Targeting. In Apoptotic Pathways as Targets for Novel Therapies in Cancer and Other Diseases; Springer: Boston, MA, USA, 2005; pp. 343–355.
- DeVita, V.T., Jr.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653.
- Guimarães, I.; dos Santos Guimarães, I.; Daltoé, R.D.; Herlinger, A.L.; Madeira, K.P.; Ladislau, T.; Valadão, I.C.; Lyra, P.C.M., Jr.; Teixeira, S.F.; Amorim, G.M.; et al. Conventional cancer treatment. In Cancer Treatment—Conventional and Innovative Approaches; Rangel, L., Ed.; Intech: Rijeka, Croatia, 2013; pp. 3–35.
- Kakde, D.; Kakde, R.; Patil, A.T.; Shrivastava, V.; Jain, D. Cancer Therapeutics-Opportunities, Challenges and Advances in Drug Delivery. J. Appl. Pharm. Sci. 2011, 1, 1–10.
- Nussbaumer, S.; Bonnabry, P.; Veuthey, J.-L.; Fleury-Souverain, S. Analysis of anticancer drugs: A review. Talanta 2011, 85, 2265–2289.
- Akhdar, H.; Legendre, C.; Aninat, C.; More, F. Anticancer Drug Metabolism: Chemotherapy Resistance and New Therapeutic Approaches. In Topics on Drug Metabolism; Paxton, J., Ed.; InTech: Rijeka, Croatia, 2012; pp. 138–170.
- Corrie, P.G. Cytotoxic chemotherapy: Clinical aspects. Medicine 2011, 39, 717–722.
- DeVita, V.T. The Evolution of Therapeutic Research in Cancer. N. Engl. J. Med. 1978, 298, 907–910.
- Osler, W. Principles Practice Medicine; D. Appleton and Company: New York, NY, USA, 1893; Volume 708.
- Fujiwara, A.; Hoshino, T.; Westley, J.W. Anthracycline Antibiotics. Crit. Rev. Biotechnol. 1985, 3, 133–157.
- Ajaykumar, C. Overview on the Side Effects of Doxorubicin. In Advances in Precision Medicine Oncology; IntechOpen: London, UK, 2020.
- Tyleckova, J.; Hrabakova, R.; Mairychova, K.; Halada, P.; Radova, L.; Džubák, P.; Hajduch, M.; Gadher, S.J.; Kovarova, H. Cancer Cell Response to Anthracyclines Effects: Mysteries of the Hidden Proteins Associated with These Drugs. Int. J. Mol. Sci. 2012, 13, 15536–15564.
- Maksimenko, A.; Dosio, F.; Mougin, J.; Ferrero, A.; Wack, S.; Reddy, L.H.; Weyn, A.A.; Lepeltier, E.; Bourgaux, C.; Stella, B.; et al. A Unique Squalenoylated and Nonpegylateddoxorubicin Nanomedicine with Systemiclong-Circulating Properties and Anticancer Activity. Proc. Natl. Acad. Sci. 2014, 111, E217–E226.
- Volkova, M. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 2012, 7, 214–220.
- Fernando, A.; Gatot, D.; Sitepu, Y.I.F. The Differences of Myelosuppression before and after Doxorubicin Chemotherapy in Breast Cancer Patients in Rsup. H. Adam Malik Medan. Int. J. Res. Rev. 2021, 8, 18–24.
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735.
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk Factors for Doxorubicin-lnduced Congestive Heart Failure. Ann. Intern. Med. 1979, 91, 710–717.
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446.
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170.
- Chang, T.M.S. Semipermeable Microcapsules. Science 1964, 146, 524–525.
- Chang, T.M.S. The in vivo Effects of Semipermeable Microcapsules containing L-Asparaginase on 6C3HED Lymphosarcoma. Nature 1971, 229, 117–118.
- Chang, T.M.S. ARTIFICIAL CELL evolves into nanomedicine, biotherapeutics, blood substitutes, drug delivery, enzyme/gene therapy, cancer therapy, cell/stem cell therapy, nanoparticles, liposomes, bioencapsulation, replicating synthetic cells, cell encapsulation/scaffold, biosorbent/immunosorbent haemoperfusion/plasmapheresis, regenerative medicine, encapsulated microbe, nanobiotechnology, nanotechnology. Artif. Cells Nanomed. Biotechnol. 2019, 47, 997–1013.
- Skipper, H.E.; Thomson, J.R.; Bell, M. Attempts at dual blocking of biochemical events in cancer chemotherapy. Cancer Res. 1954, 14, 503–507.
- Weber, G. Biochemical Strategy of Cancer Cells and the Design of Chemotherapy: G.H.A. Clowes Memorial Lecture. Cancer Res. 1983, 43, 3466–3492.
- Prabha, S.; Arya, G.; Chandra, R.; Ahmed, B.; Nimesh, S. Effect of size on biological properties of nanoparticles employed in gene delivery. Artif. Cells Nanomed. Biotechnol. 2014, 44, 83–91.
- Awad, N.S.; Paul, V.; AlSawaftah, N.M.; ter Haar, G.; Allen, T.M.; Pitt, W.G.; Husseini, G.A. Ultrasound-Responsive Nanocarriers in Cancer Treatment: A Review. ACS Pharmacol. Transl. Sci. 2021, 4, 589–612.
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics 2016, 6, 1306–1323.
- Tomao, S.; Tomao, F.; Rossi, L.; Zaccarelli, E.; Caruso, D.; Zoratto, F.; Panici, P.B.; Papa, A. Angiogenesis and antiangiogenic agents in cervical cancer. OncoTargets Ther. 2014, 7, 2237–2248.
- Dreher, M.; Liu, W.; Michelich, C.; Dewhirst, M.; Yuan, F.; Chilkoti, A. Tumor Vascular Permeability, Accumulation, and Penetration of Macromolecular Drug Carriers. J. Natl. Cancer Inst. 2006, 5, 335–344.
- Maeda, H.; Tsukigawa, K.; Fang, J. A Retrospective 30 Years After Discovery of the Enhanced Permeability and Retention Effect of Solid Tumors: Next-Generation Chemotherapeutics and Photodynamic Therapy-Problems, Solutions, and Prospects. Microcirculation 2015, 23, 173–182.
- Wang, M.; Thanou, M. Targeting nanoparticles to cancer. Pharmacol. Res. 2010, 62, 90–99.
- Al Basha, S.; Salkho, N.; Dalibalta, S.; Husseini, G.A.; Sameer, A.S.; Banday, M.Z.; Nissar, S.; Saeed, S.A. Liposomes in Active, Passive and Acoustically-Triggered Drug Delivery. Mini-Rev. Med. Chem. 2019, 19, 961–969.
- Bangham, A.; Horne, R. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668.
- Bangham, A.D.; Hill, M.W.; Miller, N.G.A. Preparation and Use of Liposomes as Models of Biological Membranes. In Methods in Membrane Biology; Korn, E., Ed.; Springer: Boston, MA, USA, 1974; pp. 1–68.
- Gregoriadis, G.; Buckland, R.A. Enzyme-containing Liposomes alleviate a Model for Storage Disease. Nature 1973, 244, 170–172.
- Cullis, P.; Mayer, L.; Bally, M.; Madden, T.; Hope, M. Generating and loading of liposomal systems for drug-delivery applications. Adv. Drug Deliv. Rev. 1989, 3, 267–282.
- Chonn, A.; Cullis, P.R. Recent advances in liposomal drug-delivery systems. Curr. Opin. Biotechnol. 1995, 6, 698–708.
- Çağdaş, M.; Sezer, A.D.; Bucak, S. Liposomes as Potential Drug Carrier Systems for Drug Delivery. In Application of Nanotechnology in Drug Delivery; Sezer, A.D., Ed.; Intech: Rijeka, Croatia, 2014.
- Crommelin, D.; van Bloois, L. Preparation and characterization of doxorubicin-containing liposomes. II. Loading capacity, long-term stability and doxorubicin-bilayer interaction mechanism. Int. J. Pharm. 1983, 17, 135–144.
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increasedin vivoactivity. Expert Opin. Drug Deliv. 2011, 8, 565–580.
- Mayer, L.; Bally, M.; Cullis, P. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochim. Biophys. Acta (BBA) Biomembr. 1986, 857, 123–126.
- Swenson, C.E.; Perkins, W.R.; Roberts, P.; Janoff, A.S. Liposome Technology and the Development of Myocet TM (Liposomal Doxorubicin Citrate). Breast 2001, 2, 1–7.
- Li, X.; Hirsh, D.J.; Cabral-Lilly, D.; Zirkel, A.; Gruner, S.M.; Janoff, A.S.; Perkins, W.R. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim. Biophys. Acta (BBA) Biomembr. 1998, 1415, 23–40.
- Kanter, P.M.; A Bullard, G.; A Ginsberg, R.; Pilkiewicz, F.G.; Mayer, L.D.; Cullis, P.R.; Pavelic, Z.P. Comparison of the cardiotoxic effects of liposomal doxorubicin (TLC D-99) versus free doxorubicin in beagle dogs. Vivo 1993, 7, 17–26.
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta (BBA) Biomembr. 1993, 1151, 201–215.
- Alyane, M.; Barratt, G.; Lahouel, M. Remote loading of doxorubicin into liposomes by transmembrane pH gradient to reduce toxicity toward H9c2 cells. Saudi Pharm. J. 2015, 24, 165–175.
- Sakakibara, T.; Chen, F.-A.; Kida, H.; Kunieda, K.; Cuenca, R.E.; Martin, F.J.; Bankert3, R.B. Doxorubicin Encapsulated in Sterically Stabilized Liposomes Is Superior to Free Drug or Drug-Containing Conventional Liposomes at Suppressing Growth and Métastases of Human Lung Tumor Xenografts1. Cancer Res. 1996, 56, 3743–3746.
- Dosio, F.; Cattel, L. PEGylation of Proteins and Liposomes: A Powerful and Flexible Strategy to Improve the Drug Delivery. Curr. Drug Metab. 2012, 13, 105–119.
- Fritze, A.; Hens, F.; Kimpfler, A.; Schubert, R.; Peschka-Süss, R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 1633–1640.
- Achim, M.; Precup, C.; Gonganău-Niţu, D.; Barbu-Tudoran, L.; Porfire, A.S.; Scurtu, R.; Ciuce, C. Thermosensitive Liposomes Containing Doxorubicin. Preparation and In Vitro Evaluation. Farmacia 2009, 57, 6.
- Pitt, W.G.; Husseini, G.A.; Roeder, B.L.; Dickinson, D.J.; Warden, D.R.; Hartley, J.M.; Jones, P.W. Preliminary Results of Combining Low Frequency Low Intensity Ultrasound and Liposomal Drug Delivery to Treat Tumors in Rats. J. Nanosci. Nanotechnol. 2011, 11, 1866–1870.
- Yuh, E.L.; Shulman, S.G.; Mehta, S.A.; Xie, J.; Chen, L.; Frenkel, V.; Bednarski, M.D.; Li, K. Delivery of Systemic Chemotherapeutic Agent to Tumors by Using Focused Ultrasound: Study in a Murine Model. Radiology 2005, 234, 431–437.
- Xu, H.; Zhang, W.; Li, Y.; Ye, F.F.; Yin, P.P.; Yu, X.; Hu, M.N.; Fu, Y.S.; Wang, C.; Shang, D.J. The Bifunctional Liposomes Constructed by Poly(2-ethyl-oxazoline)-cholesteryl Methyl Carbonate: An Effectual Approach to Enhance Liposomal Circulation Time, pH-Sensitivity and Endosomal Escape. Pharm. Res. 2014, 31, 3038–3050.
- Faraji, A.H.; Wipf, P. Nanoparticles in cellular drug delivery. Bioorganic Med. Chem. 2009, 17, 2950–2962.
- Xing, H.; Tang, L.; Yang, X.; Hwang, K.; Wang, W.; Yin, Q.; Wong, N.Y.; Dobrucki, W.; Yasui, N.; Katzenellenbogen, J.A.; et al. Selective delivery of an anticancer drug with aptamer-functionalized liposomes to breast cancer cells in vitro and in vivo. J. Mater. Chem. B 2013, 1, 5288–5297.
- Yang, B. Preclinical study of Doxorubicine-loaded liposomal drug delivery for the treatment of head and neck cancer: Optimization by Box-Behnken statistical design. Acta Biochim. Pol. 2020, 67, 149–155.
- Elamir, A.; Ajith, S.; Al Sawaftah, N.; Abuwatfa, W.; Mukhopadhyay, D.; Paul, V.; Al-Sayah, M.H.; Awad, N.; Husseini, G.A. Ultrasound-triggered herceptin liposomes for breast cancer therapy. Sci. Rep. 2021, 11, 1–13.
- Chowdhury, N.; Chaudhry, S.; Hall, N.; Olverson, G.; Zhang, Q.-J.; Mandal, T.; Dash, S.; Kundu, A. Targeted Delivery of Doxorubicin Liposomes for Her-2+ Breast Cancer Treatment. AAPS PharmSciTech 2020, 21, 202.
- Yin, X.; Feng, S.; Chi, Y.; Liu, J.; Sun, K.; Guo, C.; Wu, Z. Estrogen-functionalized liposomes grafted with glutathione-responsive sheddable chotooligosaccharides for the therapy of osteosarcoma. Drug Deliv. 2018, 25, 900–908.
- Ishida, T.; Kirchmeier, M.; Moase, E.; Zalipsky, S.; Allen, T. Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim. Biophys. Acta (BBA) Biomembr. 2001, 1515, 144–158.
- Arabi, L.; Badiee, A.; Mosaffa, F.; Jaafari, M.R. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J. Control. Release 2015, 220, 275–286.
- Wang, W.; Shao, A.; Zhang, N.; Fang, J.; Ruan, J.J.; Ruan, B.H. Cationic Polymethacrylate-Modified Liposomes Significantly Enhanced Doxorubicin Delivery and Antitumor Activity. Sci. Rep. 2017, 7, 43036.
- Ieranò, C.; Portella, L.; Lusa, S.; Salzano, G.; D’Alterio, C.; Napolitano, M.; Buoncervello, M.; Macchia, D.; Spada, M.; Barbieri, A.; et al. CXCR4-antagonist Peptide R-liposomes for combined therapy against lung metastasis. Nanoscale 2016, 8, 7562–7571.
- Li, X.; Diao, W.; Xue, H.; Wu, F.; Wang, W.; Jiang, B.; Bai, J.; Lian, B.; Feng, W.; Sun, T.; et al. Improved efficacy of doxorubicin delivery by a novel dual-ligand-modified liposome in hepatocellular carcinoma. Cancer Lett. 2020, 489, 163–173.
- Safra, T.; Muggia, F.; Jeffers, S.; Tsao-Wei, D.D.; Groshen, S.; Lyass, O.; Henderson, R.; Berry, G.; Gabizon, A. Pegylated liposomal doxorubicin (doxil): Reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann. Oncol. 2000, 11, 1029–1034.
- Kesterson, J.P.; Odunsi, K.; Lele, S. High cumulative doses of pegylated liposomal doxorubicin are not associated with cardiac toxicity in patients with gynecologic malignancies. Chemotherapy 2010, 56, 108–111.
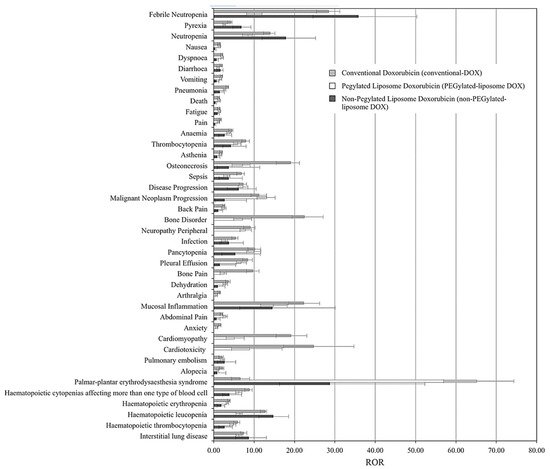
- Fukuda, A.; Tahara, K.; Hane, Y.; Matsui, T.; Sasaoka, S.; Hatahira, H.; Motooka, Y.; Hasegawa, S.; Naganuma, M.; Abe, J.; et al. Comparison of the adverse event profiles of conventional and liposomal formulations of doxorubicin using the FDA adverse event reporting system. PloS ONE 2017, 12, e0185654.



