2. Nanoparticles as Drug Delivery Systems (DDS)
Due to the detriment to the patients’ quality of life and the potential lethality of some of the side effects associated with conventional chemotherapy, novel drug delivery systems (DDS) aim to reduce the adverse side effects and enhance the specificity of chemotherapeutic drugs. In 1964, Cheng initiated the use of encapsulation of enzymes and other biologically active materials in semipermeable vesicles and tested their use to suppress the growth of lymphosarcoma in a mice model [
28,
29]. This approach has evolved and been extensively extended into nanomedicine, biotherapeutics, blood substitutes, drug delivery, enzyme/gene therapy, cancer therapy, nanoparticles, liposomes, bioencapsulation, regenerative medicine, nanobiotechnology, and nanotechnology [
30]. Earlier studies on the transformed cancer cells showed that they had adopted many biochemical strategies to support their uncontrolled growth [
31,
32]. A deep understanding of the key enzymes and antagonistic pathways of synthesis and catabolism involved in tumor progression resulted in the development of “targeted therapy”. Targeted DDS are nanoplatforms that incorporate nanoparticles (NPs) as drug delivery vehicles, ranging in size from 1 nm to 1000 nm [
33,
34]. These include, but are not limited to, gold NPs, dendrimers, polymeric nanogels, micelles, metal-organic frameworks (MOFs), liposomes, and quantum dots. The synthesis routes of these NPs vary and are generally divided into chemical and biological ways, where the latter are preferred as they are safer and innocuous [
35].
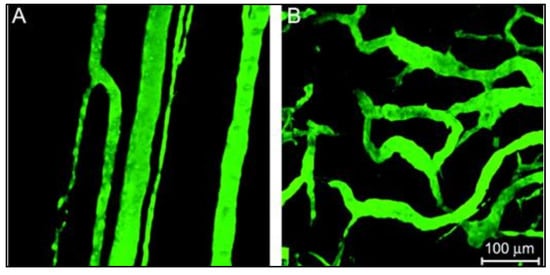
Utilizing these nanocarriers for the remote delivery of appropriate dosages to targeted anatomical sites under controlled release conditions can overcome the shortcomings of traditional treatment approaches. The localized accumulation of the nanoparticles at the neoplastic site is mainly achieved by passive and/or active targeting routes. In passive targeting, the intrinsic features of the tumor neovasculature beneficially provide fenestrations where the NPs accumulate. As a tumor undergoes rapid, chaotic growth, it exhibits a disorganized vascular network and becomes hypoxic due to insufficient oxygen supply. Tumor cells can secrete growth factors to induce vascularization to resolve this hypoxia and get nutrients from neighboring healthy cells by a process referred to as angiogenesis [
35,
36]. The newly formed capillitial endothelium in tumor tissues is disorganized and leaky with improper lymphatic drainage and inadequate transport phenomena mechanisms (
Figure 3) [
37]. Consequently, the tumor site suffers from abnormal molecular and fluid dynamics. These features allow the NPs to extravasate into the tumor’s interstitium [
38] and accumulate inside the diseased tissues, in a phenomenon referred to as the enhanced permeability and retention (EPR) effect.
Figure 3. (
A) a normal vascular network where the vessels are parallel-aligned next to each other; (
B) a tumoral vasculature with chaotic defective arrangement. Adapted with permission from [
37], Oxford University Press, 2006.
Yet, designing DDS with complete dependence on passive targeting has significant limitations, such as the possible accumulation of the NPs in the spleen and liver as these organs have fenestrated vasculature as well as the inability of the NPs to sufficiently penetrate deep enough through the complex tumoral network due to heterogeneities in structure [
39]. Thus, the development of systems that incorporate both passive and active targeting mechanisms became imperative. Since tumor cells overexpress receptors that participate in growth and survival pathways, such receptors make promising active targets. To this end, nanocarriers could be conjugated to natural ligands to target these receptors leading to their accumulation and internalization by the cancer cells [
40].
This review will discuss three types of nanocarriers: liposomes, micelles, and metal-organic frameworks (MOFs) delivering the antineoplastic agent DOX to treat different types of solid cancers, including their DOX loading techniques, encapsulation efficiency, and their ability to eradicate tumors.
3. DOX Delivery Systems Based on Liposomes
Bangham and co-workers used multilamellar bilayer lipid microspheres and liposomes as models of biological membranes for basic research in the 1960s [
41,
42]. It was Gregoriadis’ group that first used liposomes as drug carriers in the 1970s [
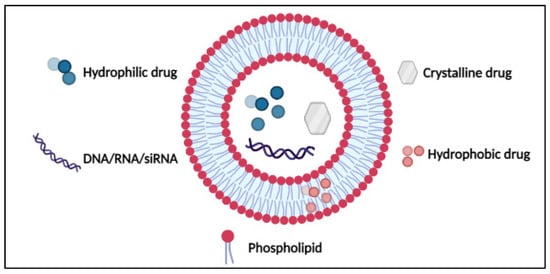
43]. Later, the multilamellar onion-like microspheres were developed into submicron dimension bilayer lipid vesicles (
Figure 4). The hydrophilic heads of the phospholipids are directed outwards, whereas the hydrophobic tails are directed inwards. Cholesterol is usually added to the phospholipids to increase the mechanical rigidity of liposomes [
44,
45]. Liposomes have gained immense interest as drug carriers because of their biocompatible, biodegradable, non-toxic, and targetable nature. Moreover, they deliver a high drug-to-lipid ratio and can also be functionalized to avoid rapid elimination from the body [
46].
Figure 4. Structure of liposomes. Reproduced from [
46], IntechOpen, 2014.
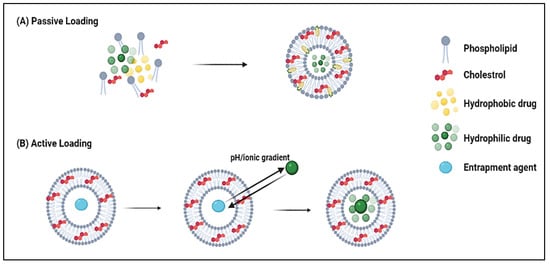
For the encapsulation of lipophilic DOX into liposomes, two loading routes (i.e., passive and active) are generally used (
Figure 5). One of the most common passive loading methods is based on the thin-film hydration method, where the drug solution is added during the liposome formation process, either while the film is being formed or during the hydration step. To enhance trapping efficiency, the drug is typically added during the film formation of liposomes containing negatively charged (acidic) lipids [
44,
45]. The dried lipids are initially randomly orientated, but upon exposure to an aqueous solution, the water causes the molecules to self-organize into a bilayer configuration to minimize entropic interactions. In the presence of hydrophilic and/or hydrophobic drugs in solution, some of the drug molecules would passively partition and accumulate in the cores and/or the shells of the liposomes. Although this method is simple, it yields very small drug encapsulation percentages. In 1983, DOX was passively entrapped in the bi-layer of positively (phosphatidylcholine-cholesterol-stearyl amine) and negatively (phosphatidylcholine-cholesterol-phosphatidylserine) charged liposomes. The drug was added to the phospholipids before film formation [
47]. One day after DOX loading, the mean diameters of positive and negative non-filtered multilamellar liposomes were between 500-600 nm and 1 μm, respectively. The maximum loading capacities ranged between 60-75 mmol DOX/mol phospholipid for the negative and approximately 55 mmol DOX/mol phospholipid for the positive, non-filtered, non-sonicated liposomes [
47].
Figure 5. Two approaches for DOX loading into liposomes: passive and active. (A) before “pasive” and (B) before “active”.
On the other hand, the size stability and release profiles were determined over an extended period. The filtered and sonicated/centrifuged negatively-charged liposomes were stable during the entire storage period and maintained average diameters of 270 and 120 nm, respectively. However, the sonicated/centrifuged positively-charged liposomes were not stable and had a size of 120 nm with a standard deviation of 40 nm over 2 weeks of storage. The negative filtered liposomes released only 10% of DOX during 75 days, whereas 40% was released over 20 days from the negative sonicated/centrifuged liposomes. For the release from positive sonicated/centrifuged liposomes, the baseline was set after 12 days, and the cumulative release was ~30% after 48 days [
47].
Other passive loading methods include the freezing-and-thawing technique (FAT). The liposomal solution undergoes a series of freezing and thawing processes, causing the interlayer distance within the liposomes to increase [
48]. As a result, ice crystals cause transient holes and pores to form within the liposomal structures, allowing for passive drug entrapment. However, this method yields 5–20% loading efficiency and requires heavy post-processing to remove the excess drug. Likewise, the reverse-evaporation method can load drugs with efficiencies up to 50%. Still, the formulations lack reliable in vivo drug retention and often suffer from rapid cargo release under physiological conditions. Thus, occurrences of “burst release”, where a large fraction of the entrapped drug is abruptly released due to poor association and retention in the vesicles, combined with the relatively low drug-to-lipid entrapment ratio, typically do not exceed 0.05% (
w/w), making it imperative to divert to other loading techniques [
48]. Moreover, the high variability and dependence of the encapsulation efficiency on operational factors like the drug’s solubility, nanocarriers’ chemistry, size, and preparation method deemed active loading methods more preferable and more commonly used for the stable entrapment of DOX into liposomes.
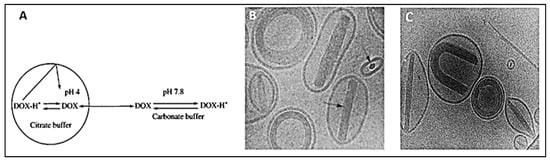
On the other hand, active methods, also known as remote loading methods, employ pH gradients between the internal acidic core buffer of blank liposomes and the external buffer containing DOX at neutral physiological conditions. Under such conditions, the drug molecules incubated with the formed liposomes can permeate selectively through the lipids transmembrane, where they become protonated intra-vesicularly [
45]. Work by Bally and colleagues [
49] investigated the effects of creating a membrane potential by altering the pH of the intraliposomal solution from a 300 mM citrate buffer at pH 4.0 to a HEPES buffer at pH 7.4 on the entrapment of DOX and biogenic amines in liposomes. High encapsulation efficiencies of up to 98% were achieved, with a drug-to-lipid ratio of 0.3 (
w/w%) corresponding to 400 nmol DOX/µmol phospholipid (~260 mM internal concentration). Also, retention times (T
50) significantly increased from 1 h to 30 h in passively versus actively DOX-loaded formulations, respectively. The proposed “citrate” method resulted in highly stable liposomes with remarkably enhanced drug retention and release characteristics, with the added benefit of cost-effectiveness. A commercially available formulation of liposomal DOX, Myocet
®, is loaded using the same approach (
Figure 6). The Myocet
® liposomes (~150 nm) are made from egg phosphatidylcholine and cholesterol at a ratio of 55/45 (mole/mole %). As a result of the pH gradient, DOX molecules diffuse and accumulate in the liposomal cores and form a complex with the citrate anions. The drug entrapment efficiencies (>95%) and drug-to-lipid ratio (~0.27) exceeded the theoretical predictions as the bundled fibers complexes allowed higher DOX loading beyond its solution solubility limits. The DOX-citrate complexes resembled the appearance of coffee-bean liposomes, where the linear bundles prevented drug leakage and premature release. The formulation also exhibited prolonged circulation times and improved in vivo tissue distribution compared to free DOX [
50]. According to Li et al. [
51], 99% of DOX loaded into citrate buffered liposomes appears in the form of fiber aggregates. The threshold for forming these fibrous-like DOX-citrate structures can be as low as ~20 mM internal DOX concentration. As the DOX concentration increases, the fibers begin cross-linking via citrates and then pack into bundles. Kanter and co-workers [
52] demonstrated that the Myocet liposomal formulation effectively reduced the effects of myocardial degeneration, commonly associated with anthracycline-based treatments. Histological analysis on beagle dogs treated with conventional DOX versus those treated with Myocet (single i.v. administration every 3 weeks; 8 cycles; cumulative dose = 12 mg/kg) showed that the latter group did not suffer from any anthracycline-induced cardiotoxicity, while the first group had lesions of myocardial degeneration.
Figure 6. (
A) DOX loading via citrate transmembrane gradient method, (
B,
C) confocal images of DOX-loaded liposomes showing the coffee-beans liposomes appearance where DOX-citrate complexes appear in rod, circular and U-shaped structures.
A,
B are adapted with permission from [
50], Elsevier, 2001. C is adapted with permission from [
51], Elsevier, 1998.
Along the same lines, Haran et al. [
53] generated a pH gradient across the liposomes using ammonium sulfate salt. The liposomes initially encapsulated a 300 mM ammonium sulfate solution at pH 5.5 in a pH 7.4 external buffer. The higher concentration of ammonium ions on the inside led to the diffusion of the neutral ammonia molecules, and with every molecule diffusing from the core, a proton was left behind. Due to salting-out effects and the acidification of the intraliposomal compartment, high fractions of DOX can accumulate in the liposomes in an aggregated form. Liposomes loaded using this technique exhibit a prolonged stable storage period beyond 6 months because of the gelation effects of DOX with the sulfate salt, which inhibit membrane re-permeation. Alyane et al. [
54] examined the stability and release behavior of liposomes loaded with DOX via the ammonium sulfate transmembrane method. Liposomes comprised of hydrogenated egg yolk phosphatidylcholine (HEPS), cholesterol, and DSPE-PEG2000 at a molar ratio of 185:1:15 showed an encapsulation efficiency exceeding 90%, with a drug-to-lipid ratio of 1:20 (
w/w). Upon incubation at 37 °C for 24 h in culture media and PBS, the liposomes retained 98% and 90% of the encapsulated drug, respectively, showing minimal leakage under the stated conditions. When the incubation medium was changed to phosphate buffer at pH 5.3, the maximum release achieved was 37%. The pH responsiveness of the liposomes suggested their suitability for controlled release in acidic compartments (i.e., tumor tissues) as they would keep their cargo intact under physiological conditions. The study also revealed that encapsulating DOX in the liposomes decreased the agent’s uptake by rat myocardial H9C2 cells, suggesting the decreased cardiac toxicity of the formulation. Flow cytometry and MTT assay analysis showed that the toxic effects of DOX were reduced when compared against that of free DOX, up to 20 h of incubation time. After 20 h, the liposomal formulation became more toxic to the cells than the free one [
54].
DOXIL
® is a commercially available PEGylated liposomal DOX formulation that uses the ammonium sulfate gradient method to encapsulate the drug. An in vivo study by Sakakibara et al. [
55] investigated the performance of DOXIL
®, conventional DOX-liposomes, and free DOX on the treatment of human lung tumors. The DOXIL
® liposomes were composed of hydrogenated soy phosphatidylcholine, cholesterol, PEG-DSPE, and dl-a-tocopherol in a molar ratio of 56.1:38.2:5.5:0.2. In contrast, the non-PEGylated liposomes had phosphatidylglycerol, phosphatidylcholine, cholesterol, and DL- α -tocopherol in a molar ratio of 1:4:3:0.02. To assess and compare the different treatment groups (dosage: 1.5 mg/kg) of anti-tumor activity, human lung tumor xenografts were engrafted into the gonadal fat pad of severe combined immunodeficient (SCID) mice. The tumor eventually metastasized from the primary site into the mice’s peritoneal cavity (i.e., liver, lung). It was concluded that the PEGylated formulation successfully suppressed the primary tumor growth and arrested metastasis in the peritoneal cavity, while the free DOX was only able to stop the growth of the primary tumor without significant effects on preventing the spread. The PEGylated liposomes showed 5-folds higher circulation times than conventional liposomes, while the uptake by vital organs like the spleen was reduced. Also, a significant increase in extravasation and accumulation of the PEGylated formulation was observed, as considerable amounts were detectable at the tumors after 1 week following administration [
55]. Another study [
56] compared the in vivo pharmacokinetic performance of free DOX with PEGylated and non-PEGylated liposomal formulations. Interestingly, the PEGylated liposomal DOX clearance rate decreased by 100-folds (Cl = 0.023 L/h), and its half-life (t1/2 = 83.7 h) was prolonged by 8-folds, compared to free DOX (Cl = 25.3 L/h, t1/2 = 10.4 h). Moreover, the distribution volume decreased significantly from 364 L to 139 L to 3.0 L in the free DOX, non-PEGylated, and PEGylated liposomal DOX, respectively. This conclusion demonstrated that PEGylation prevents premature drug release and that most of it remains entrapped without leakage.
Fritze et al. [
57] introduced another DOX remote loading method based on a phosphate ((NH
4)
2HPO
4) transmembrane gradient, using different ammonium and sodium salts. The liposomes were composed of egg phosphatidylcholine (EPC) and cholesterol in the molar ratio 7:3. After hydrating the liposomes with 300 mM salt solutions at neutral pH, they were incubated with DOX.HCl for 12 h at 7 °C to achieve a drug-to-lipids ratio of 1:3 (mol/mol). The sizes of liposomes containing DOX dissolved in different ammonium and sodium salts, as well as their encapsulation efficiencies (EE%), are summarized in
Table 1. Results showed that loading did not significantly alter the size of the liposomes, and those loaded with the ammonium salts gradients showed higher EE than the sodium salts. Synergistic effects are suggested when using ammonium salts as they act as a reservoir for donating free protons when DOX is internalized and protonated in the acidic interior of the liposomes. Thus, intraliposomal protonation and DOX precipitation led to increased encapsulation efficiencies. Further analysis was conducted to assess the impact of varying the intravesicular ammonium concentration on liposome size and EE. DOX was dissolved in 10 mM isotonic HEPES buffered saline (HBS), 50, 100, 200, and 300 mM ammonium phosphate at pH 7.2. The sizes of the DOX-loaded liposomes were found to be 84 ± 0.4, 102 ± 1.4, 114 ± 3.6, 95 ± 1.5, and 92 ± 1.6 nm, respectively, with EE of 2.81, 23.52, 61.03, 83.35, 97.98%. Increasing the ammonium ion concentration augmented the effects of drug protonation and intravesicular acidification, which led to increased DOX diffusion across the transmembrane gradient. The least efficiency (<5%) was observed when incubating in HBS, which contains no phosphate ions (no decrease in the intraliposomal pH level), while the highest efficiency approaching 100% was observed at the highest concentration of the ammonium phosphate (300 mM).
Table 1. Summary of the size and encapsulation efficiency of loaded liposomes achieved via different salts.
| Salt Gradient |
Size ± SD (nm) |
EE (%) |
| Ammonium Phosphate |
129.3 ± 3.7 |
98 |
| Ammonium Sulfate |
129.2 ± 2.9 |
95 |
| Ammonium Acetate |
115.9 ± 1.0 |
77 |
| Ammonium Citrate |
114.9 ± 1.2 |
100 |
| Sodium Phosphate |
113.4 ± 1.6 |
52 |
| Sodium Sulfate |
111.8 ± 1.9 |
44 |
| Sodium Acetate |
113.4 ± 1.6 |
16 |
| Sodium Citrate |
151.7 ± 3.8 |
54 |
Another study [
58] investigated the effect of varying the bilayer composition of DOX entrapment and release efficiency. Three liposomal formulations were tested, which included non-thermosensitive (NTS) liposomes composed of L-α-phosphatidylcholine (PC), thermosensitive (TS) liposomes composed of dipalmitoylphosphatidylcholine (DPPC) and distearoylphosphatidylcholine (DSPC), lyso-thermosensitive (LTS) liposomes composed of DPPC, DSPC, and 1-palmitoyl-2-lyso-glycero-3-phosphocholine (P-lyso-PC). All three formulations contained small amounts of cholesterol. The encapsulation efficiency of DOX dissolved in 0.9% NaCl solution was very low for TS and LTS liposomes (5.8 and 5.6%), while it was slightly higher (17.29%) for NTS liposomes because of the possibility of controlling the temperature effectively during the loading (above the phase transition temperature). In vitro drug release was modeled at controlled hyperthermia conditions (41–42 °C), and results showed that for a period of 5 h, 2%, 36%, and 54% DOX diffused from NTS, TS, and LTS liposomes. The relatively lower encapsulation efficiency and higher release performance of LTSL resort to the temperature-induced membrane instability when operating at temperatures beyond the lipids transition temperature [
58]. Generally, drug loading via active loading depends on several factors besides the drug’s physiochemical properties, like the extraliposomal medium condition, pH, conductivity, electrolytic activity, loading duration, as well as operating temperatures [
44].
Research efforts have also focused on the synergistic effects of using release triggering modalities, like ultrasound and pH, on the internalization and efficacy of DOX. Pitt et al. [
59] studied the in vivo performance of ultrasonically-triggered DOX-loaded liposomes on BDIX rats bearing colonic carcinoma. The liposomes were comprised of soy phosphatidyl choline, cholesterol, DSPE-PEG, and alpha-tocopherol combined in the molar ratio 3:1:1:0.004, and DOX was entrapped via the ammonium sulfate gradient method. Using sonication remarkably enhanced the DOX release kinetics and effects on mice. The group treated with liposomes followed by ultrasound exposure (20 kHz for 15 min, once for 4 weeks) showed significant regression in tumors growth to an immeasurable size by the end of the treatment period. Likewise, another in vivo study [
60] on mice bearing SCC7 murine squamous carcinoma cells showed that sonication using pulsed high-frequency US (HFUS) enhanced the performance of the proposed liposomal drug delivery system. Fluorescent spectrophotometry results proved that the mean DOX concentration in the sonicated tumors was 124% more than in the control, which received the liposomal treatment but without sonication. Work by Zhang et al. [
61] synthesized pH-sensitive liposomes by modifying their surfaces via the insertion of poly(2-ethyl-2-oxazoline)-cholesteryl methyl carbonate (PEOZ-CHMC) copolymer. The performance of these liposomes was compared to others grafted with PEG-DSPE chains. DOX hydrochloride was loaded into both types of liposomes by the ammonium sulfate transmembrane gradient method. It was revealed that functionalization with PEOZ-CHMC and PEG-DSPE had no effects either on the drug’s encapsulation efficiency, which was 97.3 ± 1.4, or on the size, which was ~120 nm. In vitro release done via dialysis showed that the release profile from PEOZ-CHMC-DOX liposomes in PBS at pH 5.0 significantly surpassed that observed at pH 7.4, suggesting a strong relationship between medium acidity and cargo release from the modified liposomes. Moreover, MTT assay results proved a direct relationship between the antiproliferative effects of the PEOZ-CHMC-DOX liposomes and pH conditions. The pH-sensitive formulation showed higher activity inhibition to the MCF-7 cells at lower pH; herein, pH can be used as a triggering mechanism for drug release.
As mentioned, scheming highly efficient liposomal drug delivery systems with complete dependence on passive targeting is hampered by limitations like the possible accumulation of the nanocarriers in the spleen and liver as these organs have fenestrated vasculature and their incapability to sufficiently penetrate deep enough through the complex tumoral network due to heterogeneities in structure [
62]. Benefiting from the tumor cells’ overexpression of receptors to aid in augmenting their growth and survival pathways, such receptors make promising active targets. To this end, nanocarriers can be conjugated to the ligands complementing these receptors to enhance DOX accumulation and internalization by the cancerous cells. Xing and co-workers [
63] examined the in vitro effects of functionalizing DOX-loaded PEGylated liposomes with a DNA aptamer (Apt-Urn liposomes) on MCF-7 cells. Flow cytometry results showed a 6.6-fold increase in drug uptake and efficacy of the functionalized loaded liposomes as opposed to the conventional ones, as the fluorescence response upon 4 h of incubation with the treatments corresponded to 93.6% and 57.0%, in the cells treated with Apt-Urn and control liposomes, respectively. Moreover, MTT assay results analyzed the cytotoxic effects of the Apt-Urn liposomes on cell viability. The cells were treated with different liposomal concentrations followed by an incubation period of 6 h, then re-cultured in fresh media, and the MTT test was done after 72 h. At a liposomal DOX concentration of 500 nM, cells treated with the functionalized liposomes exhibited a viability of 57.0 ± 6%, whereas cells treated with the control liposomes exhibited a viability of 92.4 ± 9%, evidencing the superior localization and internalization of the nanocarriers as a function of surface modification.
Yang [
64] conducted a recent preclinical investigation on DOX-loaded liposomes composed of EPC and cholesterol for treating head and neck cancer. The study compared three treatment groups: free DOX, DOX-loaded liposomes, and DOX-loaded peptide-conjugated liposomes. Results showed the dependence of the encapsulation efficiency on the cholesterol content in the formulations, as it increased from 20% to 79% upon the incorporation of cholesterol with EPC. Varying the EPC and cholesterol ratio yielded different entrapment efficiencies ranging from ~48% to ~82%. Both liposomal formulations, the functionalized and the conventional ones, showed sustained release profiles over a period of 40 h, but the DOX-loaded peptide-conjugated liposomes had a faster in vitro release behavior. In vivo analysis of the formulations’ efficacy was carried out on nude mice bearing HSCC (human squamous cell carcinomas) xenograft. The tumor progression was suppressed when treated with both liposomal formulations; however, the functionalized liposomes arrested metastasis and increased the median survival time of the animals in that group by 100%. The enhanced performance was due to the increased accumulation at the tumor site, resorted to localized targeting by binding to the overexpressed surface-specific receptors (Hsp47/CBP2) abundant on the head and neck cancer cells.
Another study [
65] examined the performance of DOX-loaded liposomes, functionalized with the monoclonal antibody Trastuzumab (TRA) for the enhanced targeting of breast cancer cells overexpressing the Human Epidermal growth factor Receptor 2 (HER2). The liposomes consisted of cholesterol, DPPC, and DSPE-PEG(2000)-NH
2 at a molar ratio of 30:65:5, respectively. DOX was loaded via the ammonium sulfate transmembrane gradient method, yielding a drug-to-lipids ratio of 1:6. The cargo release from the liposomal formulations, conventional and TRA-conjugated, was triggered using pulsed ultrasound. In vitro results showed synergistic effects on the DOX uptake and internalization upon sonication and surface modification with TRA. Similarly, Chowdhury and co-workers [
66] designed an in vitro study where they tested different targeted (functionalized with Aptamer-A6) and untargeted liposomal-DOX formulations for the localized treatment of HER2-overexpressing breast cancers. Twelve liposomal formulations (F1-F12) were prepared using the thin-film hydration method by varying the compositions of the phospholipid mixtures. The liposome sizes slightly increased upon DOX loading and ranged from 98.7 to 181.2 nm, with 74.9 to 94.1% entrapment efficiencies. The optimized formulations for further complexation with Aptamer-A6 were determined based on the smallest size to benefit from the EPR effect and the highest encapsulation efficiency. Formulations 5 and 8 (10 mg, 150 mg, 40 mg, 40 mg, 20 mg, and 0.25 mg of DOX, POPC, DOTAP, DOPE, DSPE-mPEG2000, and Mal-PEG, respectively) had sizes of 101.70 ± 14.04 nm and 98.7 ± 13.25 nm, respectively, and DOX entrapment efficiency of 92.8% and 94.1%, respectively. These results suggest the statistical insignificance of the hydrophobicity and charge (i.e., cationic) of the used phospholipids on the encapsulation efficiencies. However, the presence of PEG chains and the charge of the liposomal complexes directly affect their stability and integrity in long-term storage. Formulations with cationic lipids exhibited a relatively smaller size and extended shelf-life lasting up to 10 weeks, and PEG prevented liposomes from cross-linking and aggregating. Integrating Mal-PEG into the formulations aided in linking the Aptamer-A6 at its amino-terminal. In vitro analysis was done on SKBR3 and MCF7 cells, which overexpress HER2 on their surfaces. It was observed that F5 was internalized the fastest and the most by both cell lines, as flow cytometry uptake results showed the fluorescence intensities upon 2-h incubation to be 98.6% and 66.5%, respectively. SKBR3 cells uptake of F5 compared to MDA-MB-231 cells (HER2 negative cell line) was more by 1.79 times, substantiating the merit of targeted therapy.
Table 2 presents some studies involving liposomal DOX.
Table 2. Different liposomal-based DDS encapsulating DOX via the (NH4)2SO4 transmembrane gradient method.
| Preparation Method |
Target Cancer |
Functionalization |
Study Model |
Triggering Modality |
Findings |
Ref. |
| Ethanol injection |
osteosarcoma |
Estrogen |
In vitro flow cytometry and MTT analysis on MG63 (estrogen overexpressing) cells and LO2 (negative liver cells).
Ex vivo imaging of MG63 tumors extracted from Male BALB/c nude mice. |
Redox-sensitivity and glutathione responsiveness |
Loaded decorated liposomes size~110 nm.
Exhibited high encapsulation efficiency.
Ex vivo analysis of the functionalized liposomes showed more selective accumulation in tumor tissues compared to other vital organs, and in vitro results showed higher cytotoxicity towards overexpressing cells. |
[67] |
| Thin-film hydration |
Lymphoma |
anti-CD19 moiety; PEG grafted by disulfide links (mPEG-S-S-DSPE) |
In vitro MTT assay
In vivo model: Female BALB/c Cr Alt B/M mice bearing Namalwa cells |
pH sensitivity |
Liposomes decorated with cleavable PEG chains rapidly dissociated in the plasma. The pH-sensitive liposomes, targeting the CD19 epitope excessively abundant on B-lymphoma cells,
showed increased selective cytotoxicity towards these cells, and enhanced release kinetics at lower pH levels. |
[68] |
| Post-insertion; mixing with preformed DOXIL |
Cancer Stem Cells (CSCs) |
anti-CD44 monoclonal antibody (mAb) |
In vitro flow cytometry and MTT assay on C-26 and NIH-3T3 (non-tumor) cells.
In vivo model: female BALB/c mice bearing C-26 colon carcinoma. |
N/A |
Functionalization of DOXIL liposomes significantly increased their size. The IC50 values were lower on the C-26 cell line overexpressing CD44, while higher values were reported for the negative cell line (NIH-3T3). |
[69] |
| Solvent evaporation |
Various cancers |
Cationic Polymethacrylate Eudragit RL100 |
In vitro flow cytometry and MTT assay on MCF7/adr and H22 cells.
In vivo model: ICR mice bearing aggressive liver cancer H22 cells. |
N/A |
Functionalization of liposomes with Polymethacrylate derivatives increases their cellular internalization and antitumoral activity. The in vivo results showed that four injections of the functionalized formulation led to tumor size reduction by 60%. |
[70] |
| Thin-film hydration |
Metastatic lung cancer |
CXCR4-antagonist cyclic peptide (peptide R) |
In vitro cytotoxicity assay.
In vivo model: C57BL/6 mice bearing B16 human melanoma cells |
N/A |
In vitro results showed that targeting significantly decreased the IC50 while reducing metastasis and regression in tumor size growth. |
[71] |
| Film dispersion |
hepatocellular carcinoma (HCC) |
glycyrrhetinic acid (GA) and peanut agglutinin (PNA) |
In vitro specific uptake of HepG2, MCF-7, and SMMC-7721 cells
In vivo model: male BALB/C-nu mice bearing SMMC-7721 xenografts. |
N/A |
HepG2 cells showed the highest uptake towards the liposomes functionalized with GA alone, while MCF-7 showed the highest affinity towards the PNA functionalized liposomes. The dual-targeted liposomal formulation was most internalized by the SMMC-7721 |
[72] |
As many liposomal DOX formulations have successfully translated to clinical applications, and others are still in the clinical testing phase, it is essential to compare their performance and overall pharmacovigilance against the free drug.
Table 3 presents a comparison between phase III clinical trials results of two current commercially available liposomal DOX formulations, namely Myocet
® and DOXIL
®, against conventional DOX. Clinical studies have shown that DOXIL
® reduced the constraints on the dose limits of free DOX administration, in addition to reducing the cardiotoxic and myocardial damage incidences even at high doses exceeding the 500 mg/m
2 threshold [
73,
74]. Fukuda et al. [
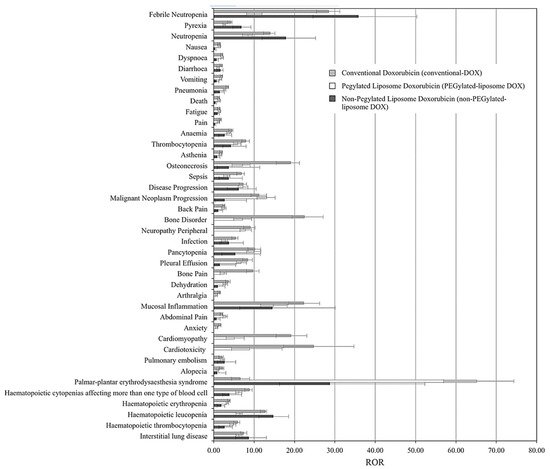
75] presented the first large-scale study that compared the adverse effects (AEs) associated with treatments of conventional, PEGylated (DOXIL
®, Caelyx
®, and LipoDox
®), and non-PEGylated (Myocet
®) liposomal DOX formulations, based on data collected from 7,561,254 patient reports (from 2004 to 2015) retrieved from the Food and Drug Administration Adverse Event Reporting System (FAERS). The analysis was based on the top 30 AEs correlated with conventional DOX treatments, including nausea, diarrhea, vomiting, anemia, cardiomyopathy, and cardiotoxicity.
Figure 7 summarizes the findings, which show the reporting odds ratios (RORs) of each treatment for each AE. ROR is a pharmacovigilance index that reflects the chances of occurrence, detection, and prevention of AEs of drug formulations. The lower the ROR, the more patient-friendly the formulation is. As reported, all DOX treatments have been correlated with severities of myelosuppression, cardiotoxicity, alopecia, nausea, and vomiting; however, both liposomal DOX formulations show relatively better safety profiles than conventional DOX.