 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tapan Kumar Mohanta | + 5130 word(s) | 5130 | 2021-11-03 04:43:57 | | | |
| 2 | Rita Xu | Meta information modification | 5130 | 2021-11-04 02:32:15 | | | | |
| 3 | Peter Tang | Meta information modification | 5130 | 2021-11-04 02:36:27 | | |
Video Upload Options
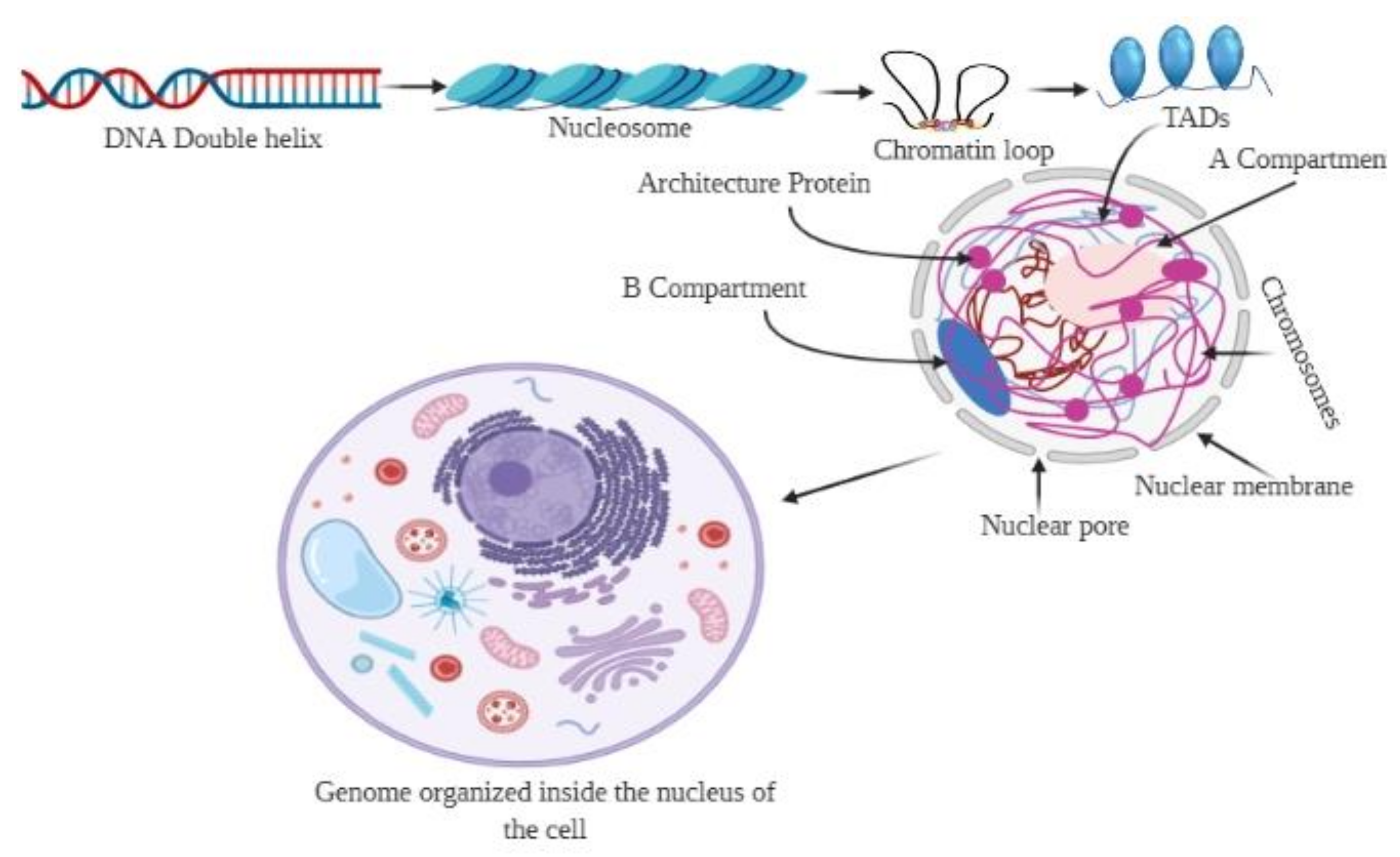
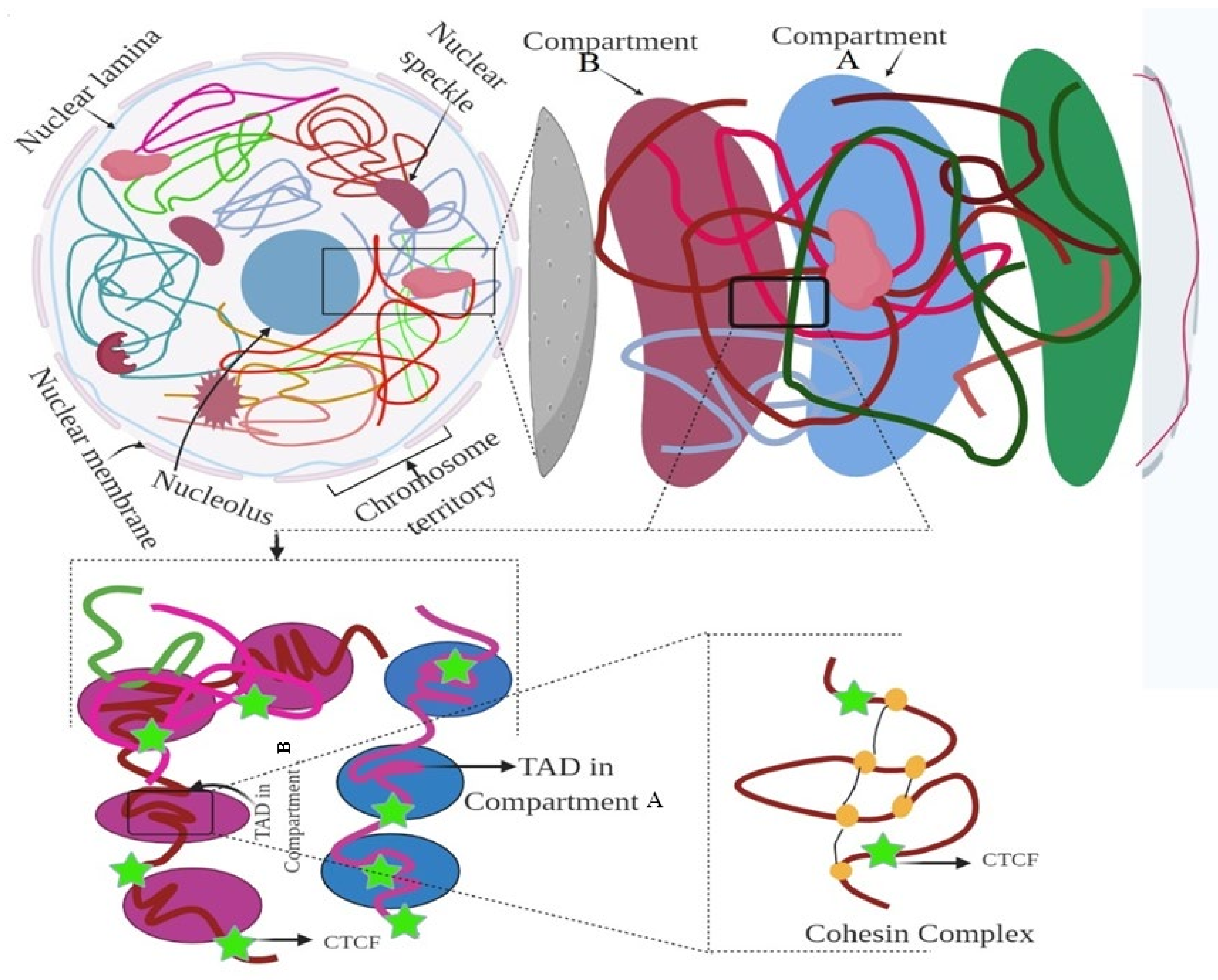
The genome is the most functional part of a cell, and genomic contents are organized in a compact three-dimensional (3D) structure. The genome contains millions of nucleotide bases organized in its proper frame. Rapid development in genome sequencing and advanced microscopy techniques have enabled us to understand the 3D spatial organization of the genome. Chromosome capture methods using a ligation approach and the visualization tool of a 3D genome browser have facilitated detailed exploration of the genome.
1. Introduction
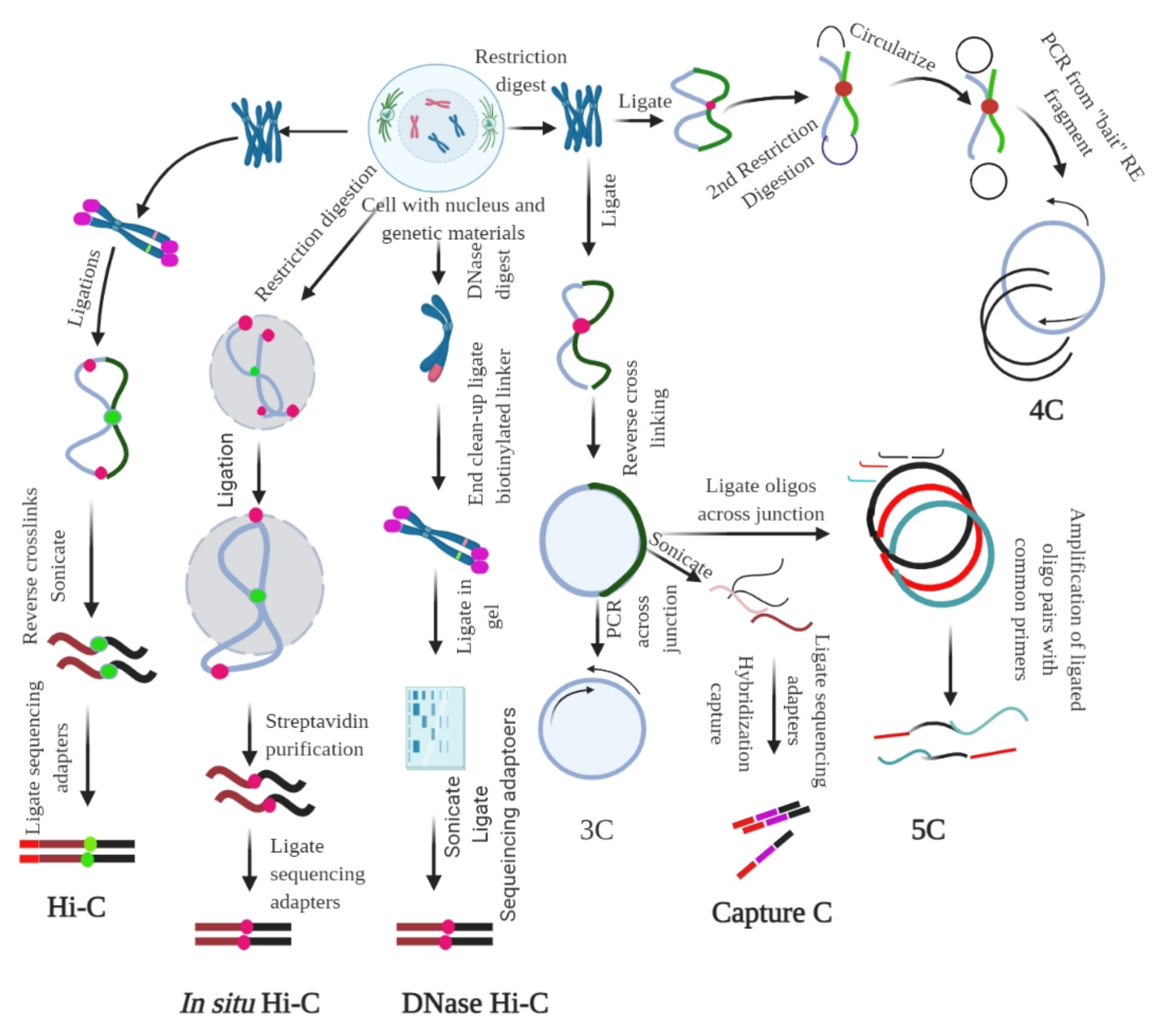
|
Method |
Assay Type |
Ligation Procedure |
Characteristics |
|---|---|---|---|
|
snHi-C |
Whole genome to whole genome |
Proximity ligation |
3C variant used to map chromatin interaction |
|
scHi-C |
Whole genome to whole genome |
Proximity ligation |
Hi-C variant enable to map chromatin interaction at single cell |
|
sciHi-C |
Whole genome to whole genome |
Proximity ligation |
Enable mapping of chromatin interactions using combinatorial barcoding |
|
3C |
One locus to one locus |
Proximity ligation |
Founding method of 3C |
|
4C |
One locus to the genome |
Proximity ligation |
Method to detect chromatin interaction between a specific locus and rest of the genome |
|
Enhanced ChIP-4C |
One to one gene |
Proximity ligation |
A variant of 4C. It improves the sensitivity through replacement of inverse PCR with primer extension |
|
Unique molecular identifier-4C |
Detect chromosomal interaction between loci and conditions |
Proximity ligation |
Improved 4C variant for improved sensitivity and specificity. It uses molecular identifier to derive high-complexity quantitative chromatin contact profiles |
|
5C |
Proximity ligation |
Method used to probe chromatin interaction of multiple loci |
|
|
CAPTURE |
One to one in the region of interest |
Proximity ligation & biotinylation |
Uses biotinylated dCas9-mediated locus specific chromatin interaction |
|
Capture-3C |
Whole genome |
Proximity ligation |
High throughput 4C that combines with 3C with DNA capture technology |
|
Capture Hi-C |
Whole genome |
Proximity ligation |
High throughput 4C that combines with Hi-C with DNA capture technology |
|
Dilution Hi-C |
Whole genome to whole genome |
Biotinylated proximity ligation |
Maps topological domains whose boarders are occupied by CTCF binding sites |
|
RNA-TRAP |
Locus to locus |
Proximity biotinylation |
Combination of RNA-FISH with ChIP to probe chromatin interaction associated with transcriptional active genes |
|
Targeted DNAse Hi-C |
Whole genome to whole genome |
Proximity ligation |
Combines DNase Hi-C with DNA capture technology |
|
Associated chromosome trap |
Long range allele specific/interchromosomal |
Proximity ligation |
Used to identify distant DNA region that interact with defined DNA target |
|
ChIA-PET |
Whole genome to whole genome mediated by protein of interest |
Proximity ligation |
Combines ChIP with proximity ligation to detect genome-wide chromatin interaction mediated by specific proteins |
|
PLAC-Seq |
Whole genome |
Proximity ligation |
Proximity ligation conducted in nuclei prior to chromatin shearing |
|
HiChIP |
Whole genome/Multi-scale |
Proximity ligation |
Combines 3C with ChIP to ascertain genome-wide chromatin interaction intervene by specific protein |
|
Hi-C |
Whole genome to whole genome |
Proximity ligation |
Used to map all chromatin interaction in a cell population |
|
DNase Hi-C |
Whole genome to whole genome |
Proximity ligation |
Is variant of Hi-C that uses DNase I to break the chromatin |
|
In Situ Hi-C |
Whole genome to whole genome |
Proximity ligation |
Is an in-situ version of Hi-C that uses chromatin digestion and proximity ligation of intact nuclei |
|
Tethered chromosome conformation capture |
Whole genome to whole genome |
Proximity ligation |
Similar to Hi-C, but ligation performed in solid substrate rather than solution |
|
In Situ DNase Hi-C |
Whole genome to whole genome |
Proximity ligation |
Hi-C variant that uses DNase to break the chromatin |
|
Micro-C |
Whole genome to whole genome |
Proximity ligation |
Is a variant of Hi-C that uses micrococcal nuclease to digest the chromatin |
|
Bridge linker Hi-C |
Whole genome |
Proximity ligation |
Used to capture structural and regulatory chromatin interaction by restriction enzymes |
|
Chromosome walks |
Whole genome |
Proximity ligation |
Links multiple genomic loci together into the proximity |
|
Genome architecture mapping (GAM) |
Whole genome |
Co-localization |
Enables identification of the interactions of enhancer and active genes across large genomic distance |
|
Split pool recognition of interaction by tag extension (SPRITE) |
Whole genome/interchromosomal |
Co-association |
Enables understanding of genome-wide detection of higher-order interactions within the nucleus |
|
Multi-ChIA |
Locus to locus |
Co-localization |
Mapping of multiplex chromatin interactions with single molecule precision. Allow mapping of chromatin interaction mediated by protein of interest |
|
Tethered conformation capture |
Chromosome scale assembly |
Proximity biotinylation |
Allows mapping of inter and intrachromosomal contacts |
2. Techniques to Study 3D Genome Organization
2.1. Microscopy-Based Visualization of the 3D Genome
2.2. Ligation-Based Detection of Contacts
2.3. Non-Ligation-Based Detection
2.4. Cell Imaging of the Nuclear Structure
3. Hierarchy of the 3D Genome
4. 3D Genome and Gene Expression
5. Data Structure of the 3D Genome
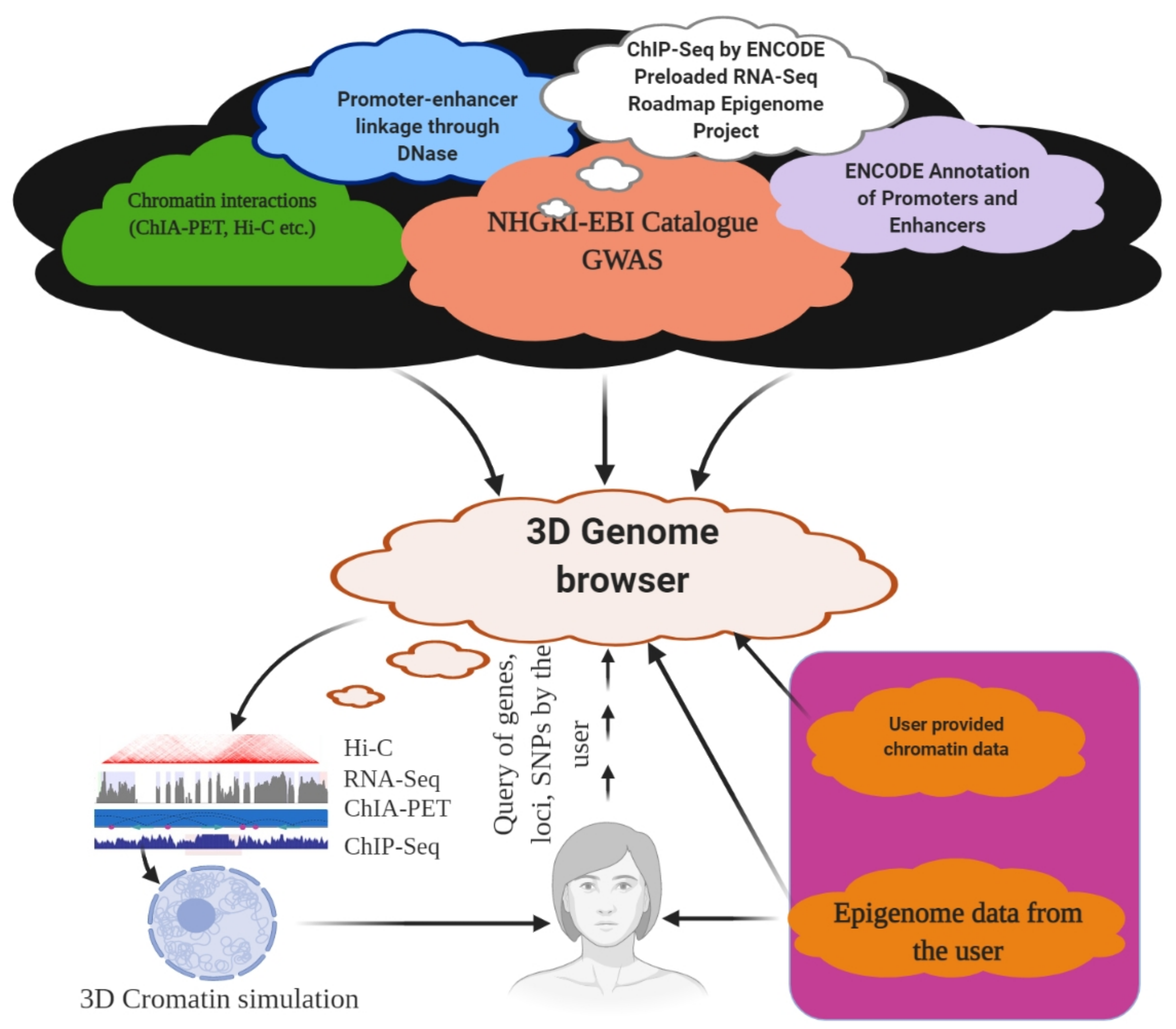
6. 3D Genome Browser
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351.
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433.
- Church, D.M.; Schneider, V.A.; Graves, T.; Auger, K.; Cunningham, F.; Bouk, N.; Chen, H.-C.; Agarwala, R.; McLaren, W.M.; Ritchie, G.R.S.; et al. Modernizing Reference Genome Assemblies. PLoS Biol. 2011, 9, e1001091.
- Tian, Y.; Zeng, Y.; Zhang, J.; Yang, C.; Yan, L.; Wang, X.; Shi, C.; Xie, J.; Dai, T.; Peng, L.; et al. High quality reference genome of drumstick tree (Moringa oleifera Lam.), a potential perennial crop. Sci. China Life Sci. 2015, 58, 627–638.
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465.
- Razin, S.V.; Farrell, C.M.; Recillas-Targa, F.B.T.-I.R. Genomic Domains and Regulatory Elements Operating at the Domain Level. Int. Rev. Cytol. 2003, 226, 63–125.
- Bernardi, G. THE HUMAN GENOME: Organization and Evolutionary History. Annu. Rev. Genet. 1995, 29, 445–476.
- Parada, L.A.; Sotiriou, S.; Misteli, T. Spatial genome organization. Exp. Cell Res. 2004, 296, 64–70.
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550.
- Rowley, M.J.; Nichols, M.H.; Lyu, X.; Ando-Kuri, M.; Rivera, I.S.M.; Hermetz, K.; Wang, P.; Ruan, Y.; Corces, V.G. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 2017, 67, 837–852.e7.
- Wang, S.; Xu, J.; Zeng, J. Inferential modeling of 3D chromatin structure. Nucleic Acids Res. 2015, 43, e54.
- Tjio, J.H.; Levan, A. The Chromosome Number of Man. In Problems of Birth Defects: From Hippocrates to Thalidomide and After; Persaud, T.V.N., Ed.; Springer: Dordrecht, The Netherlands, 1977; pp. 112–118. ISBN 978-94-011-6621-8.
- Felker, P.; Paterson, A.; Jenderek, M.M. Forage potential of Opuntia clones maintained by the USDA, National Plant Germplasm System (NPGS) collection. Crop Sci. 2006, 46, 2161–2168.
- Breidenbach, N.; Sharov, V.V.; Gailing, O.; Krutovsky, K.V. De novo transcriptome assembly of cold stressed clones of the hexaploid Sequoia sempervirens (D. Don) Endl. Sci. Data 2020, 7, 239.
- Cenci, A.; Combes, M.-C.; Lashermes, P. Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant Mol. Biol. 2012, 78, 135–145.
- Sinha, B.M.B.; Srivastava, D.P.; Jha, J. Occurrence of Various Cytotypes of Ophioglossum reticulatum L. in a Population from N. E. India. Caryologia 1979, 32, 135–146.
- Swart, E.C.; Bracht, J.R.; Magrini, V.; Minx, P.; Chen, X.; Zhou, Y.; Khurana, J.S.; Goldman, A.D.; Nowacki, M.; Schotanus, K.; et al. The Oxytricha trifallax Macronuclear Genome: A Complex Eukaryotic Genome with 16,000 Tiny Chromosomes. PLoS Biol. 2013, 11, e1001473.
- Kempfer, R.; Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2020, 21, 207–226.
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668.
- Kim, S.; Yu, N.-K.; Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 2015, 47, e166.
- Krijger, P.H.L.; de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016, 17, 771–782.
- Cubeñas-Potts, C.; Corces, V.G. Architectural proteins, transcription, and the three-dimensional organization of the genome. FEBS Lett. 2015, 589, 2923–2930.
- Todolli, S.; Perez, P.J.; Clauvelin, N.; Olson, W.K. Contributions of Sequence to the Higher-Order Structures of DNA. Biophys. J. 2017, 112, 416–426.
- Zhao, Y.; Zhan, Q. Electric oscillation and coupling of chromatin regulate chromosome packaging and transcription in eukaryotic cells. Theor. Biol. Med. Model. 2012, 9, 27.
- Gavrilov, A.A.; Shevelyov, Y.Y.; Ulianov, S.V.; Khrameeva, E.E.; Kos, P.; Chertovich, A.; Razin, S. V Unraveling the mechanisms of chromatin fibril packaging. Nucleus 2016, 7, 319–324.
- Luzhin, A.V.; Flyamer, I.M.; Khrameeva, E.E.; Ulianov, S.V.; Razin, S.V.; Gavrilov, A.A. Quantitative differences in TAD border strength underly the TAD hierarchy in Drosophila chromosomes. J. Cell. Biochem. 2019, 120, 4494–4503.
- Jakoby, M.; Schnittger, A. Cell cycle and differentiation. Curr. Opin. Plant Biol. 2004, 7, 661–669.
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678.
- Kong, S.; Zhang, Y. Deciphering Hi-C: From 3D genome to function. Cell Biol. Toxicol. 2019, 35, 15–32.
- Eichler, E.E.; Sankoff, D. Structural Dynamics of Eukaryotic Chromosome Evolution. Science 2003, 301, 793–797.
- Chen, Z.J. Genetic and Epigenetic Mechanisms for Gene Expression and Phenotypic Variation in Plant Polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406.
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627.
- Uhler, C.; Shivashankar, G. V Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727.
- Starr, D.A.; Fridolfsson, H.N. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 2010, 26, 421–444.
- Li, Q.; Kumar, A.; Makhija, E.; Shivashankar, G. V The regulation of dynamic mechanical coupling between actin cytoskeleton and nucleus by matrix geometry. Biomaterials 2014, 35, 961–969.
- Maniotis, A.J.; Chen, C.S.; Ingber, D.E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA 1997, 94, 849–854.
- Hakim, O.; Misteli, T. SnapShot: Chromosome confirmation capture. Cell 2012, 148, 1068.e1–1068.e2.
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing Chromosome Conformation. Science 2002, 295, 1306–1311.
- De Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24.
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, 1869.
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276.
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64.
- Melo, U.S.; Schöpflin, R.; Acuna-Hidalgo, R.; Mensah, M.A.; Fischer-Zirnsak, B.; Holtgrewe, M.; Klever, M.-K.; Türkmen, S.; Heinrich, V.; Pluym, I.D.; et al. Hi-C Identifies Complex Genomic Rearrangements and TAD-Shuffling in Developmental Diseases. Am. J. Hum. Genet. 2020, 106, 872–884.
- Giorgetti, L.; Heard, E. Closing the loop: 3C versus DNA FISH. Genome Biol. 2016, 17, 215.
- Bolland, D.J.; King, M.R.; Reik, W.; Corcoran, A.E.; Krueger, C. Robust 3D DNA FISH using directly labeled probes. J. Vis. Exp. 2013, 50587.
- Übelmesser, N.; Papantonis, A. Technologies to study spatial genome organization: Beyond 3C. Brief. Funct. Genomics 2019, 18, 395–401.
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2018, 9, 20–32.
- Kaul, A.; Bhattacharyya, S.; Ay, F. Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat. Protoc. 2020, 15, 991–1012.
- Eijsbouts, C.Q.; Burren, O.S.; Newcombe, P.J.; Wallace, C. Fine mapping chromatin contacts in capture Hi-C data. BMC Genomics 2019, 20, 77.
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.A.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; de Santiago, I.; Lavitas, L.-M.; Branco, M.R.; et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 2017, 543, 519–524.
- Liu, T.; Wang, Z. normGAM: An R package to remove systematic biases in genome architecture mapping data. BMC Genomics 2019, 20, 1006.
- Le Treut, G.; Képès, F.; Orland, H. A Polymer Model for the Quantitative Reconstruction of Chromosome Architecture from HiC and GAM Data. Biophys. J. 2018, 115, 2286–2294.
- Rusk, N. SPRITE maps the 3D genome. Nat. Methods 2018, 15, 572.
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e24.
- Koch, L. Getting the drop on chromatin interaction. Nat. Rev. Genet. 2019, 20, 192–193.
- Zheng, M.; Tian, S.Z.; Capurso, D.; Kim, M.; Maurya, R.; Lee, B.; Piecuch, E.; Gong, L.; Zhu, J.J.; Li, Z.; et al. Multiplex chromatin interactions with single-molecule precision. Nature 2019, 566, 558–562.
- Risca, V.I.; Greenleaf, W.J. Unraveling the 3D genome: Genomics tools for multiscale exploration. Trends Genet. 2015, 31, 357–372.
- Schwarzacher, T.; Anamthawat-Jónsson, K.; Harrison, G.E.; Islam, A.K.M.R.; Jia, J.Z.; King, I.P.; Leitch, A.R.; Miller, T.E.; Reader, S.M.; Rogers, W.J.; et al. Genomic in situ hybridization to identify alien chromosomes and chromosome segments in wheat. Theor. Appl. Genet. 1992, 84, 778–786.
- Fukui, K.; Ohmido, N.; Khush, G.S. Variability in rDNA loci in the genus Oryza detected through fluorescence in situ hybridization. Theor. Appl. Genet. 1994, 87, 893–899.
- Trouvelot, S.; van Tuinen, D.; Hijri, M.; Gianinazzi-Pearson, V. Visualization of ribosomal DNA loci in spore interphasic nuclei of glomalean fungi by fluorescence in situ hybridization. Mycorrhiza 1999, 8, 203–206.
- Shakoori, A.R. Fluorescence In Situ Hybridization (FISH) and Its Applications. Chromosom. Struct. Aberrations 2017, 343–367.
- Cui, C.; Shu, W.; Li, P. Fluorescence In situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89.
- Bishop, R. Applications of fluorescence in situ hybridization (FISH) in detecting genetic aberrations of medical significance. Biosci. Horizons Int. J. Student Res. 2010, 3, 85–95.
- Safak, Y.L.; Daniel, R.N. Mechanistic Approach to the Problem of Hybridization Efficiency in Fluorescent In Situ Hybridization. Appl. Environ. Microbiol. 2004, 70, 7126–7139.
- Solanki, S.; Ameen, G.; Zhao, J.; Flaten, J.; Borowicz, P.; Brueggeman, R.S. Visualization of spatial gene expression in plants by modified RNAscope fluorescent in situ hybridization. Plant Methods 2020, 16, 1–9.
- Diot, C.; Chin, A.; Lécuyer, E. Optimized FISH methods for visualizing RNA localization properties in Drosophila and human tissues and cultured cells. Methods 2017, 126, 156–165.
- Johnson, M.D., III; Fresco, J.R. Third-strand in situ hybridization (TISH) to non-denatured metaphase spreads and interphase nuclei. Chromosoma 1999, 108, 181–189.
- Egozcue, J.; Blanco, J.; Vidal, F. Chromosome studies in human sperm nuclei using fluorescence in-situ hybridization (FISH). Hum. Reprod. Update 1997, 3, 441–452.
- Alamri, A.; Nam, J.Y.; Blancato, J.K. Fluorescence In Situ Hybridization of Cells, Chromosomes, and Formalin-Fixed Paraffin-Embedded Tissues. In Molecular Profiling: Methods and Protocols; Espina, V., Ed.; Springer: New York, NY, USA, 2017; pp. 265–279. ISBN 978-1-4939-6990-6.
- Solovei, I.; Cavallo, A.; Schermelleh, L.; Jaunin, F.; Scasselati, C.; Cmarko, D.; Cremer, C.; Fakan, S.; Cremer, T. Spatial Preservation of Nuclear Chromatin Architecture during Three-Dimensional Fluorescence in Situ Hybridization (3D-FISH). Exp. Cell Res. 2002, 276, 10–23.
- Fields, B.D.; Nguyen, S.C.; Nir, G.; Kennedy, S. A multiplexed DNA FISH strategy for assessing genome architecture in Caenorhabditis elegans. eLife 2019, 8, e42823.
- Karafiátová, M.; Bartoš, J.; Kopecký, D.; Ma, L.; Sato, K.; Houben, A.; Stein, N.; Doležel, J. Mapping nonrecombining regions in barley using multicolor FISH. Chromosom. Res. 2013, 21, 739–751.
- Moore, L.E.; Titenko-Holland, N.; Quintana, P.J.E.; Smith, M.T. Novel biomarkers of genetic damage in humans: Use of fluorescence in situ hybridization to detect aneuploidy and micronuclei in exfoliated cells. J. Toxicol. Environ. Health 1993, 40, 349–357.
- Madon, P.F.; Athalye, A.S.; Sanghavi, K.; Parikh, F.R. Microdeletion Syndromes Detected by FISH–73 Positive from 374 Cases. Int. J. Hum. Genet. 2010, 10, 15–20.
- ALmughamsi, M.M.; Kumosani, T.A.; ALhamzi, E.A.; Al-Qahtani, M. The use of fluorescence in situhybridization techniques in the detection of microdeletion syndromes. BMC Genomics 2014, 15, P60.
- Masny, P.S.; Chan, O.Y.A.; de Greef, J.C.; Bengtsson, U.; Ehrlich, M.; Tawil, R.; Lock, L.F.; Hewitt, J.E.; Stocksdale, J.; Martin, J.H.; et al. Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur. J. Hum. Genet. 2010, 18, 448–456.
- Rosin, L.F.; Gil, J., Jr.; Drinnenberg, I.A.; Lei, E.P. Oligopaint DNA FISH reveals telomere-based meiotic pairing dynamics in the silkworm, Bombyx mori. PLoS Genet. 2021, 17, e1009700.
- Beliveau, B.J.; Kishi, J.Y.; Nir, G.; Sasaki, H.M.; Saka, S.K.; Nguyen, S.C.; Wu, C.; Yin, P. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc. Natl. Acad. Sci. USA 2018, 115, E2183–E2192.
- Torres-Ruiz, R.; Grazioso, T.P.; Brandt, M.; Martinez-Lage, M.; Rodriguez-Perales, S.; Djouder, N. Detection of chromosome instability by interphase FISH in mouse and human tissues. STAR Protoc. 2021, 2, 100631.
- Boyle, S.; Rodesch, M.J.; Halvensleben, H.A.; Jeddeloh, J.A.; Bickmore, W.A. Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosom. Res. 2011, 19, 901–909.
- Hans de Jong, J.; Fransz, P.; Zabel, P. High resolution FISH in plants—Techniques and applications. Trends Plant Sci. 1999, 4, 258–263.
- Gudla, P.R.; Nakayama, K.; Pegoraro, G.; Misteli, T. SpotLearn: Convolutional Neural Network for Detection of Fluorescence In Situ Hybridization (FISH) Signals in High-Throughput Imaging Approaches. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 57–70.
- Mayer, R.; Brero, A.; von Hase, J.; Schroeder, T.; Cremer, T.; Dietzel, S. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 2005, 6, 44.
- Xie, S.Q.; Lavitas, L.-M.; Pombo, A. CryoFISH: Fluorescence In Situ Hybridization on Ultrathin Cryosections. In iFluorescence in situ Hybridization (FISH): Protocols and Applications; Bridger, J.M., Volpi, E.V., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 219–230. ISBN 978-1-60761-789-1.
- Branco, M.R.; Pombo, A. Intermingling of Chromosome Territories in Interphase Suggests Role in Translocations and Transcription-Dependent Associations. PLoS Biol. 2006, 4, e138.
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet. 2006, 38, 1348–1354.
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306.
- Beliveau, B.J.; Apostolopoulos, N.; Wu, C. Visualizing Genomes with Oligopaint FISH Probes. Curr. Protoc. Mol. Biol. 2014, 105, 14.23.1–14.23.20.
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189.
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422.
- Ni, Y.; Cao, B.; Ma, T.; Niu, G.; Huo, Y.; Huang, J.; Chen, D.; Liu, Y.; Yu, B.; Zhang, M.Q.; et al. Super-resolution imaging of a 2.5 kb non-repetitive DNA in situ in the nuclear genome using molecular beacon probes. eLife 2017, 6, e21660.
- Di Stefano, M.; Di Giovanni, F.; Pozharskaia, V.; Gomar-Alba, M.; Baù, D.; Carey, L.B.; Marti-Renom, M.A.; Mendoza, M. Impact of Chromosome Fusions on 3D Genome Organization and Gene Expression in Budding Yeast. Genetics 2020, 214, 651–667.
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA replication timing in the 3D genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737.
- Smyk, M.; Szafranski, P.; Startek, M.; Gambin, A.; Stankiewicz, P. Chromosome conformation capture-on-chip analysis of long-range cis-interactions of the SOX9 promoter. Chromosom. Res. 2013, 21, 781–788.
- Van de Werken, H.J.G.; Landan, G.; Holwerda, S.J.B.; Hoichman, M.; Klous, P.; Chachik, R.; Splinter, E.; Valdes-Quezada, C.; Öz, Y.; Bouwman, B.A.M.; et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 2012, 9, 969–972.
- Loviglio, M.N.; Leleu, M.; Männik, K.; Passeggeri, M.; Giannuzzi, G.; van der Werf, I.; Waszak, S.M.; Zazhytska, M.; Roberts-Caldeira, I.; Gheldof, N.; et al. Chromosomal contacts connect loci associated with autism, BMI and head circumference phenotypes. Mol. Psychiatry 2017, 22, 836–849.
- Dear, P.H.; Cook, P.R. Happy mapping: A proposal for linkage mapping the human genome. Nucleic Acids Res. 1989, 17, 6795–6807.
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64.
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A pathway for mitotic chromosome formation. Science 2018, 359, eaao6135.
- Bickmore, W.A. The Spatial Organization of the Human Genome. Annu. Rev. Genomics Hum. Genet. 2013, 14, 67–84.
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 2013, 155, 1479–1491.
- Fudenberg, G.; Kelley, D.R.; Pollard, K.S. Predicting 3D genome folding from DNA sequence with Akita. Nat. Methods 2020, 17, 1111–1117.
- Mullinger, A.M.; Johnson, R.T. Packing DNA into chromosomes. J. Cell Sci. 1980, 46, 61–86.
- Luger, K.; Hansen, J.C. Nucleosome and chromatin fiber dynamics. Curr. Opin. Struct. Biol. 2005, 15, 188–196.
- Belmont, A.S.; Sedat, J.W.; Agard, D.A. A three-dimensional approach to mitotic chromosome structure: Evidence for a complex hierarchical organization. J. Cell Biol. 1987, 105, 77–92.
- Gibcus, J.H.; Dekker, J. The Hierarchy of the 3D Genome. Mol. Cell 2013, 49, 773–782.
- Dorier, J.; Stasiak, A. Topological origins of chromosomal territories. Nucleic Acids Res. 2009, 37, 6316–6322.
- Spilianakis, C.G.; Lalioti, M.D.; Town, T.; Lee, G.R.; Flavell, R.A. Interchromosomal associations between alternatively expressed loci. Nature 2005, 435, 637–645.
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293.
- Dekker, J.; Misteli, T. Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 2015, 7, a019356.
- Mekhail, K.; Moazed, D. The nuclear envelope in genome organization, expression and stability. Nat. Rev. Mol. Cell Biol. 2010, 11, 317–328.
- Van de Vosse, D.W.; Wan, Y.; Wozniak, R.W.; Aitchison, J.D. Role of the nuclear envelope in genome organization and gene expression. WIREs Syst. Biol. Med. 2011, 3, 147–166.
- Kinney, N.A.; Onufriev, A.V.; Sharakhov, I. V Quantified effects of chromosome-nuclear envelope attachments on 3D organization of chromosomes. Nucleus 2015, 6, 212–224.
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326.
- Broers, J.L.V.; Kuijpers, H.J.H.; Östlund, C.; Worman, H.J.; Endert, J.; Ramaekers, F.C.S. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp. Cell Res. 2005, 304, 582–592.
- Briand, N.; Collas, P. Lamina-associated domains: Peripheral matters and internal affairs. Genome Biol. 2020, 21, 85.
- Van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791.
- Maji, A.; Ahmed, J.A.; Roy, S.; Chakrabarti, B.; Mitra, M.K. A Lamin-Associated Chromatin Model for Chromosome Organization. Biophys. J. 2020, 118, 3041–3050.
- Towbin, B.D.; Gonzalez-Sandoval, A.; Gasser, S.M. Mechanisms of heterochromatin subnuclear localization. Trends Biochem. Sci. 2013, 38, 356–363.
- Mattout, A.; Cabianca, D.S.; Gasser, S.M. Chromatin states and nuclear organization in development—A view from the nuclear lamina. Genome Biol. 2015, 16, 174.
- Imai, S.; Nishibayashi, S.; Takao, K.; Tomifuji, M.; Fujino, T.; Hasegawa, M.; Takano, T. Dissociation of Oct-1 from the Nuclear Peripheral Structure Induces the Cellular Aging-associated Collagenase Gene Expression. Mol. Biol. Cell 1997, 8, 2407–2419.
- Lanctôt, C.; Cheutin, T.; Cremer, M.; Cavalli, G.; Cremer, T. Dynamic genome architecture in the nuclear space: Regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007, 8, 104–115.
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417.
- Finlan, L.E.; Sproul, D.; Thomson, I.; Boyle, S.; Kerr, E.; Perry, P.; Ylstra, B.; Chubb, J.R.; Bickmore, W.A. Recruitment to the Nuclear Periphery Can Alter Expression of Genes in Human Cells. PLoS Genet. 2008, 4, e1000039.
- Lemaître, C.; Bickmore, W.A. Chromatin at the nuclear periphery and the regulation of genome functions. Histochem. Cell Biol. 2015, 144, 111–122.
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31.
- Karoutas, A.; Akhtar, A. Functional mechanisms and abnormalities of the nuclear lamina. Nat. Cell Biol. 2021, 23, 116–126.
- Bridger, J.M.; Foeger, N.; Kill, I.R.; Herrmann, H. The nuclear lamina. FEBS J. 2007, 274, 1354–1361.
- Pepenella, S.; Hayes, J. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes: Commentary. Chemtracts 2007, 20, 406–409.
- Ptak, C.; Aitchison, J.D.; Wozniak, R.W. The multifunctional nuclear pore complex: A platform for controlling gene expression. Curr. Opin. Cell Biol. 2014, 28, 46–53.
- Németh, A.; Längst, G. Genome organization in and around the nucleolus. Trends Genet. 2011, 27, 149–156.
- Pontvianne, F.; Carpentier, M.-C.; Durut, N.; Pavlištová, V.; Jaške, K.; Schořová, Š.; Parrinello, H.; Rohmer, M.; Pikaard, C.S.; Fojtová, M.; et al. Identification of Nucleolus-Associated Chromatin Domains Reveals a Role for the Nucleolus in 3D Organization of the A. thaliana Genome. Cell Rep. 2016, 16, 1574–1587.
- Németh, A.; Conesa, A.; Santoyo-Lopez, J.; Medina, I.; Montaner, D.; Péterfia, B.; Solovei, I.; Cremer, T.; Dopazo, J.; Längst, G. Initial Genomics of the Human Nucleolus. PLoS Genet. 2010, 6, e1000889.
- Therizols, P.; Duong, T.; Dujon, B.; Zimmer, C.; Fabre, E. Chromosome arm length and nuclear constraints determine the dynamic relationship of yeast subtelomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 2025–2030.
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.S. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367.
- Meaburn, K.J.; Misteli, T. Chromosome territories. Nature 2007, 445, 379–381.
- Wang, Y.; Botvinick, E.L.; Zhao, Y.; Berns, M.W.; Usami, S.; Tsien, R.Y.; Chien, S. Visualizing the mechanical activation of Src. Nature 2005, 434, 1040–1045.
- Le, H.Q.; Ghatak, S.; Yeung, C.-Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875.
- Nakazawa, N.; Sathe, A.R.; Shivashankar, G.V.; Sheetz, M.P. Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. Proc. Natl. Acad. Sci. USA 2016, 113, E6813–E6822.
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G. V Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11349–11354.
- Chen, C.S.; Mrksich, M.; Huang, S.; Whitesides, G.M.; Ingber, D.E. Geometric Control of Cell Life and Death. Science 1997, 276, 1425–1428.
- Kilian, K.A.; Bugarija, B.; Lahn, B.T.; Mrksich, M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4872–4877.
- Thomas, C.H.; Collier, J.H.; Sfeir, C.S.; Healy, K.E. Engineering gene expression and protein synthesis by modulation of nuclear shape. Proc. Natl. Acad. Sci. USA 2002, 99, 1972–1977.
- Suter, D.M.; Molina, N.; Gatfield, D.; Schneider, K.; Schibler, U.; Naef, F. Mammalian Genes Are Transcribed with Widely Different Bursting Kinetics. Science 2011, 332, 472–474.
- Bartman, C.R.; Hamagami, N.; Keller, C.A.; Giardine, B.; Hardison, R.C.; Blobel, G.A.; Raj, A. Transcriptional Burst Initiation and Polymerase Pause Release Are Key Control Points of Transcriptional Regulation. Mol. Cell 2019, 73, 519–532.e4.
- Chen, H.; Levo, M.; Barinov, L.; Fujioka, M.; Jaynes, J.B.; Gregor, T. Dynamic interplay between enhancer–promoter topology and gene activity. Nat. Genet. 2018, 50, 1296–1303.
- Cho, W.-K.; Jayanth, N.; English, B.P.; Inoue, T.; Andrews, J.O.; Conway, W.; Grimm, J.B.; Spille, J.-H.; Lavis, L.D.; Lionnet, T.; et al. RNA Polymerase II cluster dynamics predict mRNA output in living cells. eLife 2016, 5, e13617.
- Fanucchi, S.; Shibayama, Y.; Burd, S.; Weinberg, M.S.; Mhlanga, M.M. Chromosomal Contact Permits Transcription between Coregulated Genes. Cell 2013, 155, 606–620.
- Noordermeer, D.; Leleu, M.; Schorderet, P.; Joye, E.; Chabaud, F.; Duboule, D. Temporal dynamics and developmental memory of 3D chromatin architecture at Hox gene loci. eLife 2014, 3, e02557.
- Letsou, W.; Cai, L. Noncommutative Biology: Sequential Regulation of Complex Networks. PLoS Comput. Biol. 2016, 12, e1005089.
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375.
- Hatch, E.; Hetzer, M. Breaching the nuclear envelope in development and disease. J. Cell Biol. 2014, 205, 133–141.
- Finch, J.T.; Klug, A. Solenoidal model for superstructure in chromatin. Proc. Natl. Acad. Sci. USA 1976, 73, 1897–1901.
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025.
- Banigan, E.J.; Mirny, L.A. Loop extrusion: Theory meets single-molecule experiments. Curr. Opin. Cell Biol. 2020, 64, 124–138.
- Terakawa, T.; Bisht, S.; Eeftens, J.M.; Dekker, C.; Haering, C.H.; Greene, E.C. The condensin complex is a mechanochemical motor that translocates along DNA. Science 2017, 358, 672–676.
- Kelman, Z.; O’Donnell, M. DNA polymerase III holoenzyme: Structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 1995, 64, 171–200.
- Wang, M.D.; Schnitzer, M.J.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Force and Velocity Measured for Single Molecules of RNA Polymerase. Science 1998, 282, 902–907.
- Bintu, B.; Mateo, L.J.; Su, J.-H.; Sinnott-Armstrong, N.A.; Parker, M.; Kinrot, S.; Yamaya, K.; Boettiger, A.N.; Zhuang, X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 2018, 362, eaau1783.
- Szabo, Q.; Jost, D.; Chang, J.-M.; Cattoni, D.I.; Papadopoulos, G.L.; Bonev, B.; Sexton, T.; Gurgo, J.; Jacquier, C.; Nollmann, M.; et al. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018, 4, eaar8082.
- Huang, K.; Li, Y.; Shim, A.R.; Nap, R.J.; Agrawal, V.; Virk, R.K.A.; Eshein, A.; Almassalha, L.M.; Backman, V.; Szleifer, I. Physical and data structure of 3D genome. Sci. Adv. 2020, 6, eaay4055.
- Rabin, A.; Neer, A. Crumpled Globule Model of the Three-Dimensional Structure of DNA. EPL (Europhys. Lett.) 2006, 23, 373.
- Mirny, L.A. The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 2011, 19, 37–51.
- Bianco, S.; Chiariello, A.M.; Conte, M.; Esposito, A.; Fiorillo, L.; Musella, F.; Nicodemi, M. Computational approaches from polymer physics to investigate chromatin folding. Curr. Opin. Cell Biol. 2020, 64, 10–17.
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226.
- Nicodemi, M.; Prisco, A. Thermodynamic Pathways to Genome Spatial Organization in the Cell Nucleus. Biophys. J. 2009, 96, 2168–2177.
- Jost, D.; Carrivain, P.; Cavalli, G.; Vaillant, C. Modeling epigenome folding: Formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res. 2014, 42, 9553–9561.
- Chiariello, A.M.; Annunziatella, C.; Bianco, S.; Esposito, A.; Nicodemi, M. Polymer physics of chromosome large-scale 3D organisation. Sci. Rep. 2016, 6, 29775.
- Bianco, S.; Lupiáñez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schöpflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat. Genet. 2018, 50, 662–667.
- Brackley, C.A.; Johnson, J.; Kelly, S.; Cook, P.R.; Marenduzzo, D. Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucleic Acids Res. 2016, 44, 3503–3512.
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophys. J. 2018, 114, 2262–2270.
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28.
- Zhou, X.; Maricque, B.; Xie, M.; Li, D.; Sundaram, V.; Martin, E.A.; Koebbe, B.C.; Nielsen, C.; Hirst, M.; Farnham, P.; et al. The Human Epigenome Browser at Washington University. Nat. Methods 2011, 8, 989–990.
- Zhou, X.; Lowdon, R.F.; Li, D.; Lawson, H.A.; Madden, P.A.F.; Costello, J.F.; Wang, T. Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods 2013, 10, 375–376.
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101.
- Kerpedjiev, P.; Abdennur, N.; Lekschas, F.; McCallum, C.; Dinkla, K.; Strobelt, H.; Luber, J.M.; Ouellette, S.B.; Azhir, A.; Kumar, N.; et al. HiGlass: Web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018, 19, 125.
- Tang, B.; Li, F.; Li, J.; Zhao, W.; Zhang, Z. Delta: A new web-based 3D genome visualization and analysis platform. Bioinformatics 2018, 34, 1409–1410.
- Wang, Y.; Song, F.; Zhang, B.; Zhang, L.; Xu, J.; Kuang, D.; Li, D.; Choudhary, M.N.K.; Li, Y.; Hu, M.; et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018, 19, 151.

