 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ik-Soo Lee | + 2912 word(s) | 2912 | 2021-07-02 11:31:01 | | | |
| 2 | Amina Yu | Meta information modification | 2912 | 2021-07-12 05:01:29 | | |
Video Upload Options
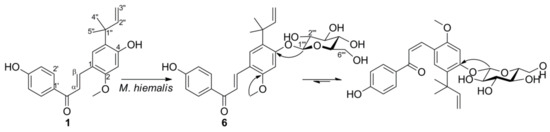
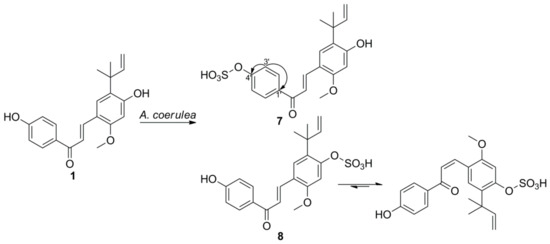
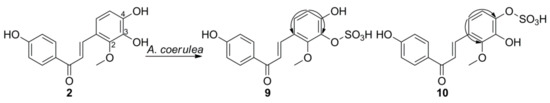
Microbial conjugation studies of licochalcones (1–4) and xanthohumol (5) were performed by using the fungi Mucor hiemalis and Absidia coerulea. As a result, one new glucosylated metabolite was produced by M. hiemalis whereas four new and three known sulfated metabolites were obtained by transformation with A. coerulea. Chemical structures of all the metabolites were elucidated on the basis of 1D-, 2D-NMR and mass spectroscopic data analyses. These results could contribute to a better understanding of the metabolic fates of licochalcones and xanthohumol in mammalian systems. Although licochalcone A 4′-sulfate (7) showed less cytotoxic activity against human cancer cell lines compared to its substrate licochalcone A, its activity was fairly retained with the IC50 values in the range of 27.35–43.07 μM.
1. Introduction
2. Microbial Transformation of Licochalcone A and Xanthohumol by M. hiemalis
3. Microbial Transformation of Licochalcones A, B, C, H, and Xanthohumol by A. coerulea
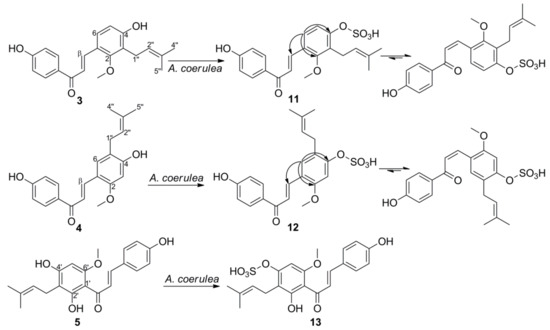
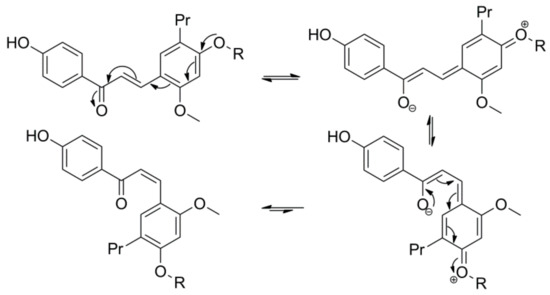
| Compound | Cell Lines (IC50, μM) | ||
|---|---|---|---|
| A375P | MCF-7 | A549 | |
| 1 | 12.86 ± 3.42 | 19.16 ± 0.65 | 18.14 ± 1.26 |
| 5 | 11.02 ± 2.09 | 23.11 ± 1.36 | 26.93 ± 1.56 |
| 7 | 27.35 ± 1.44 | 30.85 ± 4.58 | 43.07 ± 1.63 |
| 13 | 41.04 ± 1.61 | 81.27 ± 3.05 | 99.40 ± 1.99 |
| DZ 1 | 9.86 ± 0.57 | 5.12 ± 0.44 | 4.01 ± 0.78 |
1 Used as positive control.
References
- Maria Pia, G.D.; Sara, F.; Mario, F.; Lorenza, S. Biological effects of licochalcones. Mini Rev. Med. Chem. 2019, 19, 647–656.
- Wang, C.; Chen, L.; Xu, C.; Shi, J.; Chen, S.; Tan, M.; Chen, J.; Zou, L.; Chen, C.; Liu, Z.; et al. A comprehensive review for phytochemical, pharmacological, and biosynthesis studies on Glycyrrhiza spp. Am. J. Chin. Med. 2020, 48, 1–29.
- Weng, Q.; Chen, L.; Ye, L.; Lu, X.; Yu, Z.; Wen, C.; Chen, Y.; Huang, G. Determination of licochalcone A in rat plasma by UPLC-MS/MS and its pharmacokinetics. Acta Chromatogr. 2019, 31, 262–265.
- Nadelmann, L.; Tjørnelund, J.; Hansen, S.H.; Cornett, C.; Sidelmann, U.G.; Braumann, U.; Christensen, E.; Christensen, S.B. Synthesis, isolation and identification of glucuronides and mercapturic acids of a novel antiparasitic agent, licochalcone A. Xenobiotica 1997, 27, 667–680.
- Huang, L.; Nikolic, D.; van Breemen, R.B. Hepatic metabolism of licochalcone A, a potential chemopreventive chalcone from licorice (Glycyrrhiza inflata), determined using liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 6937–6948.
- Nadelmann, L.; Tjørnelund, J.; Christensen, E.; Hansen, S.H. High-performance liquid chromatographic determination of licochalcone A and its metabolites in biological fluids. J. Chromatogr. B Biomed. Sci. Appl. 1997, 695, 389–400.
- Yilmazer, M.; Stevens, J.F.; Buhler, D.R. In vitro glucuronidation of xanthohumol, a flavonoid in hop and beer, by rat and human liver microsomes. FEBS Lett. 2001, 491, 252–256.
- Yilmazer, M.; Stevens, J.F.; Deinzer, M.L.; Buhler, D.R. In vitro biotransformation of xanthohumol, a flavonoid from hops (Humulus lupulus), by rat liver microsomes. Drug Metab. Dispos. 2001, 29, 223–231.
- Nikolic, D.; Li, Y.; Chadwick, L.R.; Pauli, G.F.; van Breemen, R.B. Metabolism of xanthohumol and isoxanthohumol, prenylated flavonoids from hops (Humulus lupulus L.) by human liver microsomes. J. Mass Spectrom. 2005, 40, 289–299.
- Ruefer, C.E.; Gerhäuser, C.; Frank, N.; Becker, H.; Kulling, S.E. In vitro phase II metabolism of xanthohumol by human UDP-glucuronosyltransferases and sulfotransferases. Mol. Nutr. Food Res. 2005, 49, 851–856.
- Jirásko, R.; Holčapek, M.; Vrublová, E.; Ulrichová, J.; Šimánek, V. Identification of new phase II metabolites of xanthohumol in rat in vivo biotransformation of hop extracts using high-performance liquid chromatography electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 4100–4108.
- Böhmdorfer, M.; Szakmary, A.; Schiestl, R.H.; Vaquero, J.; Riha, J.; Brenner, S.; Thalhammer, T.; Szekeres, T.; Jäger, W. Involvement of UDP-glucuronosyltransferases and sulfotransferases in the excretion and tissue distribution of resveratrol in mice. Nutrients 2017, 9, 1347.
- Marhol, P.; Hartog, A.F.; van der Horst, M.A.; Wever, R.; Purchartová, K.; Fuksová, K.; Kuzma, M.; Cvačka, J.; Křen, V. Preparation of silybin and isosilybin sulfates by sulfotransferase from Desulfitobacterium hafniense. J. Mol. Catal. B. Enzym. 2013, 89, 24–27.
- Sak, K. Chapter 6-Anticancer action of sulfated flavonoids as phase II metabolites. In Handbook of Food Bioengineering, Food Bioconversion; Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: Cambridge, MA, USA; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; Volume 2, pp. 207–236.
- Koizumi, M.; Shimizu, M.; Kobashi, K. Enzymatic sulfation of quercetin by arylsulfotransferase from a human intestinal bacterium. Chem. Pharm. Bull. 1990, 38, 794–796.
- Correia-da-Silva, M.; Sousa, E.; Pinto, M.M.M. Emerging sulfated flavonoids and other polyphenols as drugs: Nature as an inspiration. Med. Res. Rev. 2014, 34, 223–279.
- Roubalová, L.; Purchartová, K.; Papoušková, K.; Vacek, J.; Křen, V.; Ulrichová, J.; Vrba, J. Sulfation modulates the cell uptake, antiradical activity and biological effects of flavonoids in vitro: An examination of quercetin, isoquercitrin and taxifolin. Bioorg. Med. Chem. 2015, 23, 5402–5409.
- Teles, Y.C.F.; Souza, M.S.R.; de Souza, M.F.V. Sulphated flavonoids: Biosynthesis, structures, and biological activities. Molecules 2018, 23, 480.
- Arunrattiyakorn, P.; Suksamrarn, S.; Suwannasai, N.; Kanzaki, H. Microbial metabolism of α-mangostin isolated from Garcinia mangostana L. Phytochemistry 2011, 72, 730–734.
- Xiao, Y.; Han, F.; Lee, I.-S. Microbial transformation of licochalcones. Molecules 2020, 25, 60.
- Kim, H.J.; Lee, I.-S. Microbial metabolism of the prenylated chalcone xanthohumol. J. Nat. Prod. 2006, 69, 1522–1524.
- Feng, J.; Zhang, P.; Cui, Y.; Li, K.; Qiao, X.; Zhang, Y.T.; Li, S.M.; Cox, R.J.; Wu, B.; Ye, M.; et al. Regio- and stereospecific O-glycosyation of phenolic compounds catalyzed by a fungal glycosyltransferases from Mucor hiemalis. Adv. Synth. Catal. 2017, 359, 995–1006.
- Han, F.; Lee, I.-S. A new flavonol glycoside from the aerial parts of Epimedium koreanum Nakai. Nat. Prod. Res. 2017, 33, 320–325.
- Ibrahim, A.-R.S. Sulfation of naringenin by Cunninghamella elegans. Phytochemistry 2000, 53, 209–212.
- Ibrahim, A.-R.S.; Galal, A.M.; Ahmed, M.S.; Mossa, G.S. O-Demethylation and sulfation of 7-methoxylated flavonones by Cunninghamella elegans. Chem. Pharm. Bull. 2003, 51, 203–206.
- Kajiyama, K.; Demizu, S.; Hiraga, Y.; Kinoshita, K.; Koyama, K.; Takahashi, K.; Tamura, Y.; Okada, K.; Kinoshita, T. Two prenylated retrochalcones from Glycyrrhiza inflata. Phytochemistry 1992, 31, 3229–3232.
- Wang, Z.; Cao, Y.; Paudel, S.; Yoon, G.; Cheon, S.H. Concise synthesis of licochalcone C and its regioisomer, licochalcone H. Arch. Pharm. Res. 2013, 36, 4132–4136.
- Chen, Q.-H.; Fu, M.-L.; Chen, M.-M.; Liu, J.; Liu, X.-J.; He, G.-Q.; Pu, S.-C. Preparative isolation and purification of xanthohumol from hops (Humulus lupulus L.) by high-speed counter-current chromatography. Food Chem. 2012, 132, 619–623.
- Basílio, N.; Bittar, S.A.; Mora, N.; Dangles, O.; Pina, F. Analogs of natural 3-deoxyanthocyanins: O-glucosides of the 4′,7-dihydroxyflavylium ion and the deep influence of glycosidation on color. Int. J. Mol. Sci. 2016, 17, 1751.
- Simmler, C.; Lankin, D.C.; Nikolić, D.; van Breemen, R.B.; Pauli, G.F. Isolation and structural characterization of dihydrobenzofuran congers of licochalcone A. Fitoterapia 2017, 121, 6–15.
- Correia, M.S.P.; Lin, W.; Aria, A.J.; Jain, A.; Globisch, D. Rapid preparation of a large sulfated metabolite library for structure validation in human samples. Metabolites 2020, 10, 415.
- Van der Horst, M.A.; Hartog, A.F.; El Morabet, R.; Marais, A.; Kircz, M.; Wever, R. Enzymatic sulfation of phenolic hydroxy groups of various plant metabolites by an arylsulfotransferase. Eur. J. Org. Chem. 2015, 2015, 534–541.
- Herath, W.; Mikell, J.R.; Hale, A.L.; Ferreira, D.; Khan, I.A. Microbial metabolism part 9. Structure and antioxidant significance of the metabolites of 5,7-dihydroxyflavone (chrysin), and 5- and 6-hydroxyflavones. Chem. Pharm. Bull. 2008, 56, 418–422.
- Mikell, J.R.; Khan, I.A. Bioconversion of 7-hydroxyflavanone: Isolation, characterization and bioactivity evaluation of twenty-one phase I and phase II microbial metabolites. Chem. Pharm. Bull. 2012, 60, 1139–1145.
- Ibrahim, A.K.; Radwan, M.M.; Ahmed, S.A.; Slade, D.; Ross, S.A.; ElSohly, M.A.; Khan, I.A. Microbial metabolism of cannflavin A and B isolated from Cannabis sativa. Phytochemistry 2010, 71, 1014–1019.
- Mikell, J.R.; Herath, W.; Khan, I.A. Microbial metabolism. Part 12. Isolation, characterization and bioactivity evaluation of eighteen microbial metabolites of 4′-hydroxyflavanone. Chem. Pharm. Bull. 2011, 59, 692–697.
- Herath, W.; Khan, I.A. Microbial metabolism. Part 13. Metabolites of hesperetin. Bioorg. Med. Chem. Lett. 2011, 21, 5784–5786.
- Bartmańska, A.; Tronina, T.; Huszcza, E. Transformation of 8-prenylnaringenin by Absidia coerulea and Beauveria bassiana. Bioorg. Med. Chem. Lett. 2012, 22, 6451–6453.
- Fan, N.; Du, C.H.; Xu, J.Q.; Xu, Y.X.; Yu, B.Y.; Zhang, J. Glycosylation and sulfation of 4-methylumbelliferone by Gliocladium deliquescens NRRL 1086. Appl. Biochem. Microbiol. 2017, 53, 85–93.
- Feng, J.; Liang, W.; Ji, S.; Qiao, X.; Zhang, Y.; Yu, S.; Ye, M. Microbial transformation of isoangustone A by Mucor hiemalis CGMCC 3.14114. J. Chin. Pharm. Sci. 2015, 24, 285–291.
- Ruan, F.; Chen, R.; Li, J.; Zhang, M.; Xie, K.; Wang, Y.; Feng, R.; Dai, J. Sulfation of naringenin by Mucor sp. Zhongguo Zhongyao Zazhi 2014, 39, 2039–2042.
- Bartmańska, A.; Tronina, T.; Huszcza, E. Microbial sulfation of 8-prenylnaringenin. Z. Naturforsch 2013, 68c, 231–235.
- Yuan, W.; Zhang, L.-P.; Cheng, K.-D.; Zhu, P.; Wang, Q.; He, H.-X.; Zhu, H.-X. Microbial O-demethylation, hydroxylation, sulfation, and ribosylation of a xanthone derivative from Halenia elliptica. J. Nat. Prod. 2006, 69, 811–814.
- Xie, L.; Xiao, D.; Wang, X.; Wang, C.; Bai, J.; Yue, Q.; Yue, H.; Li, Y.; Molnár, I.; Xu, Y.; et al. Combinatorial biosynthesis of sulfated benzenediol lactones with a phenolic sulfotransferase from Fusarium graminearum PH-1. mSphere 2020, 5, e00949-20.
- Wang, P.; Yuan, X.; Wang, Y.; Zhao, H.; Sun, X.; Zheng, Q. Licochalcone C induces apoptosis via B-cell lymphoma 2 family proteins in T24 cells. Mol. Med. Rep. 2015, 12, 7623–7628.
- Liu, Y.; Wang, Y.; Yan, X.; Si, L.; Cao, C.; Yu, L.; Zheng, Q. Anti-proliferation effects of licochalcone B on human breast cancer MCF-7 cells. Zhongguo Shiyan Fangjixue Zazhi 2016, 22, 106–111.
- Kang, T.H.; Yoon, G.; Kang, I.-A.; Oh, H.-N.; Chae, J.-I.; Shim, J.-H. Natural compound licochalcone B induced extrinsic and intrinsic apoptosis in human skin melanoma (A375) and Squamous cell carcinoma (A431) cells. Phytother. Res. 2017, 31, 1858–1867.
- Cho, J.J.; Chae, J.-I.; Yoon, G.; Ki, M.K.H.; Cho, J.H.; Cho, S.-S.; Cho, Y.S.; Shim, J.-H. Licochalcone A, a natural chalconoid isolated from Glycyrrhiza inflata root, induces apoptosis via Sp1 and Sp1 regulatory proteins in oral squamous cell carcinoma. Int. J. Oncol. 2014, 45, 667–674.
- Oh, H.-N.; Yoon, G.; Shin, J.-C.; Park, S.-M.; Cho, S.-S.; Cho, J.H.; Lee, M.-H.; Liu, K.D.; Cho, Y.S.; Chae, J.-I.; et al. Licochalcone B induces apoptosis of human oral squamous cell carcinoma through the extrinsic- and intrinsic-signaling pathways. Int. J. Oncol. 2016, 48, 1749–1757.
- Oh, H.-N.; Seo, J.-H.; Lee, M.-H.; Kim, C.; Kim, E.; Yoon, G.; Cho, S.-S.; Cho, Y.S.; Choi, H.W.; Shim, J.-H.; et al. Licochalcone C induces apoptosis in human oral squamous cell carcinoma cells by regulation of the JAK2/STAT3 signaling pathway. J. Cell. Biochem. 2018, 119, 10118–10130.
- Oh, H.-N.; Oh, K.B.; Lee, M.-H.; Seo, J.-H.; Kim, E.; Yoon, G.; Cho, S.-S.; Cho, Y.S.; Choi, H.W.; Chae, J.-I.; et al. JAK2 regulation by licochalcone H inhibits the cell growth and induces apoptosis in human oral squamous cell carcinoma. Phytomedicine 2019, 52, 60–69.
- Szliszka, E.; Czuba, Z.P.; Mazur, B.; Sedek, L.; Paradysz, A.; Krol, W. Chalcones enhance TRAIL-induced apoptosis in prostate cancer cells. Int. J. Mol. Sci. 2010, 11, 1–13.

