 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hoon Hur | + 4079 word(s) | 4079 | 2021-04-20 09:44:46 | | | |
| 2 | Peter Tang | Meta information modification | 4079 | 2021-06-10 03:32:27 | | |
Video Upload Options
Gastrointestinal (GI) cancers are primary malignant tumors associated with cancer-related deaths worldwide. Although chemotherapy and radiotherapy are essential modalities to improve patient survival, many patients show resistance to these therapies. Various clinical studies have suggested that cancer-associated fibroblasts (CAFs) play a significant role in this resistance.
1. Introduction
Cancers originating from the gastrointestinal (GI) tract, including the esophagus, stomach, colorectum, liver, and pancreas, are common malignancies and are the primary cause of cancer-related mortalities worldwide [1]. The core treatment strategy for GI cancers is surgical resection. However, patients with non-resectable or recurrent disease are predominantly treated with chemotherapeutic agents or radiation techniques as a palliative measure [2]. Targeting agents and immunotherapy are recently developed alternatives for improving the survival of GI cancer patients [3]. However, most patients with advanced-stage GI cancers are resistant to these treatment modalities; thus, their survival rates remain dismal.
Several studies have investigated the mechanisms underlying resistance to therapy in cancers originating from the GI tract. These studies have focused on the tumor cells themselves, such as drug efflux through transmembrane transport proteins and anti-apoptotic protein activation [4][5]. However, to date, agents that block these pathways have not yet been applied in clinical settings. Moreover, numerous studies have revealed that the tumor microenvironment (TME) may play a pivotal role in resistance to chemotherapy and radiotherapy [6][7]. The TME of solid cancers comprises various non-cancerous cells, the extracellular matrix, and soluble factors [8][9] that enhance tumorigenesis, invasion, metastasis, and therapy resistance in cancer cells. Therefore, targeting agents that block the interaction between cancer cells and the TME may improve treatment outcomes in GI cancer patients [10].
Cancer-associated fibroblasts (CAFs) constitute a significant component of the TME in GI cancers. They are involved in cancer invasion and tumor growth through their interaction with cancer cells and immune microenvironments [11][12]. Numerous studies have reported that CAFs can trigger the resistance of cancer cells to treatments [13][14][15][16]. Therefore, CAFs have emerged as a novel treatment target to improve the efficacy of chemotherapy and radiotherapy in GI cancers. However, drugs targeting CAFs have not yet been administered to patients.
2. Clinical Evidence for the Role of CAFs in Chemotherapy and Radiotherapy Resistance in GI Cancer
The desmoplastic reaction developed by the recruited fibroblasts is prominently observed in progressed GI cancers [17], and this reaction has been considered a major cause of resistance to chemoradiotherapy [18]. Some clinical studies have demonstrated that high desmoplasia is significantly correlated with poor clinical outcomes in patients with GI cancers, such as pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC) [19][20][21]. Therefore, treatment strategies targeting tumor desmoplasia have mainly tried to improve the survival of patients with advanced GI cancers [11]. For example, the monoclonal antibody for fibroblast activation protein (anti-FAP mAb) showed some therapeutic effects in CRC without severe toxicity in the early phase of a clinical trial [22]. Additionally, recent phase II clinical trials testing angiotensin I receptor blockers as inhibitors of CAF activation and pegvorhyaluronidase alfa as a decomposer of hyaluronan accumulated by CAFs; these trials have described improved outcomes in PDAC patients [23][24]. Although these agents have not yet been approved as a treatment of choice for GI cancers, accumulating evidence suggests that targeting CAFs in GI cancers is promising.
It has been confirmed through immunohistochemistry (IHC) that CAF accumulation in GI cancers is related to chemotherapy resistance. Ma et al. performed IHC for alpha-smooth muscle actin (α-SMA) in paraffin-embedded formalin-fixed (PEFF) tissues of gastric cancer (GC) patients treated with chemotherapy. The results showed that the GC tissues of patients showing resistance to chemotherapy contained more α-SMA-positive CAFs than the chemosensitive patients [25]. Other CRC studies also reported a significant correlation between a high proportion of α-SMA-expressing CAFs and resistance to 5-fluorouracil plus oxaliplatin-based chemotherapy [26].
The expression of CAF-derived molecules in human GI cancer tissues could be investigated to provide clinical evidence. Some researchers have reported a direct correlation between biomarkers originating in stromal cells and response to neoadjuvant treatment. The expression of the two markers FAP-α and C-X-C motif chemokine ligand (CXCL) 12, known as stromal cell-derived factor 1 (SDF-1), was positively associated with poor clinical outcomes in rectal cancer patients who underwent neoadjuvant chemoradiation [27][28]. Although chemoradiotherapy is the most popular modality for esophageal cancers (ESOCs), patients have frequently exhibited resistance to therapies, resulting in poor outcomes [29]. One study described the expression of CXCL1 in ESOC tissue specimens biopsied after chemoradiation. We concluded that the upregulation of CXCL1 in CAFs was an independent prognostic factor in these patients [30]. In addition, positive transforming growth factor-beta (TGF-β) expression in CAFs of ESOC tissues was significantly correlated with poor survival outcomes in patients treated with chemoradiotherapy [31]. Another group reported that high PAI-1 expression in CAFs led to considerably worse progression-free survival in ESOC patients treated with cisplatin [32].
Large-scale cancer genome studies using high-throughput technologies have provided comprehensive molecular profiling information for solid cancers [33]. The Cancer Genome Atlas (TCGA) consortium has suggested molecular subgroups and treatment targets based on a genome-scale analysis using bulk tumors of large cohorts [34][35][36][37][38]. However, considering the role of non-cancerous cells in the bulk tumors on cancer progression and therapeutic efficacy, the meanings of these cell fractions should be investigated. Algorithms including ESTIMATE [39], CIBERSORT [40], EPIC [41], and MCP-counter [42] can predict the proportion of stromal or immune cells in bulk cancer tissues. Consequently, the implications of the accumulation of these cells in GI cancer patient prognosis can be inferred. Recent high-throughput transcriptome analyses of GI cancers have highlighted that stroma-related genes have unfavorable outcomes in patients with various types of GI cancers, including GC, CRC, PDAC, and hepatocellular carcinoma [43][44][45][46][47]. However, these results were obtained using surgical specimens from patients who underwent curative resection, with or without subsequent adjuvant systemic treatment. To define the correlation between gene expression and response to chemotherapy, expression analyses in pretreated samples from patients subjected to preoperative chemotherapy can undoubtedly reflect their responsiveness to chemotherapy based on gene expression. Our recent data obtained using NanoString transcriptome analysis revealed that stroma-related gene expression in pretreated endoscopic biopsy tissues of GC patients who underwent preoperative chemotherapy significantly correlated with an inadequate response to chemotherapy [15]. Although NanoString transcriptome analysis screens a limited number of genes, it could be applied to a small number of samples, such as endoscopic biopsy specimens. The results implied that the high expression of stroma-related genes in the biopsied tissues of GC patients might require a novel treatment strategy. This strategy may improve the efficacy of chemotherapy for patients with GC (Figure 1). However, our study had some limitations, including a low number of enrolled patients. Another study with a large number of GC patients who had undergone neoadjuvant treatment showed that several genetic mutations could serve as predictive markers for chemotherapy response [48]. However, future studies investigating TMEs should be conducted to assess their role in therapy resistance.
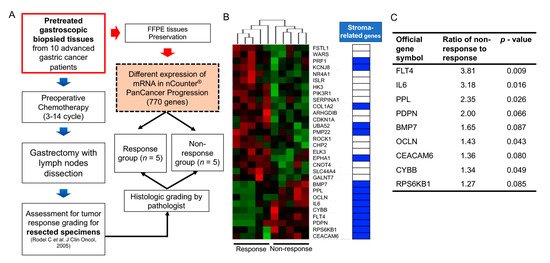
Collectively, these findings indicate that CAF accumulation or CAF-specific markers in malignant tumors originating from the GI tract are significantly related to chemotherapy or radiotherapy resistance. Therefore, the mechanisms underlying the interaction between CAFs and cancer cells would act as excellent targets to improve the responsiveness of GI cancer patients to chemoradiotherapy.
3. Origin of CAFs in GI Cancer
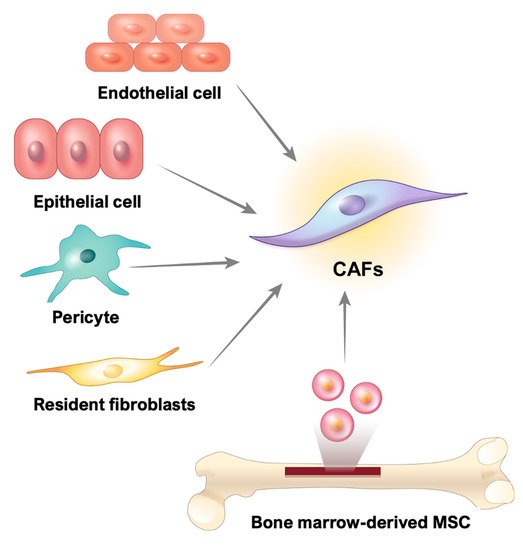
CAFs are fibrotic cells involved in tumor malignancy; however, the origin of these cells in GI cancer remains unclear. Numerous studies have reported that CAFs may be derived from resident fibroblasts, smooth muscle cells, endothelial cells, pericytes, bone marrow-derived stem cells, and even epithelial cells [49][50] (Figure 2).
Genetic and functional comparisons between fibroblasts isolated from surgically resected cancers and paired healthy tissues are relatively easier to perform than those isolated from other cell types; therefore, resident fibroblasts have been extensively explored in this context [51]. The unique characteristics of CAFs compared to normal resident fibroblasts could indicate the potential mechanism underlying the transdifferentiation of normal fibroblasts to CAFs in GI cancers. TGF-β is produced by colon cancer cells and activates the differentiation of residual colon fibroblasts into CAFs during colon cancer progression. These activated CAFs upregulate the expression of activated markers, such as α-SMA and FAPs, and produce large amounts of glycoproteins, including tenascin-C and collagen maturation enzymes, for extracellular matrix (ECM) remodeling [52]. GC cells of the scirrhous subtype also produce TGF-β, which indicates the expression of α-SMA in normal residual fibroblasts [53]. Moreover, the aforementioned study proposed that TGF-β could be reciprocally involved in the CAF-induced stemness of scirrhous GC cells and demonstrated that anti-TGF-β antibody had an inhibitory effect on tumor growth.
In PDAC, one of the distinct origins of CAFs may be pancreatic stellate cells (PSCs), the resident mesenchymal cells of the noncancerous pancreas [54]. Similar to GC, PDAC cell-induced TGF-β can activate PSCs and increase the production of ECM components such as fibronectin, collagen, and tenascin-C [55]. Additionally, the sonic hedgehog (SHH) protein expressed in PDAC cells contributes to tumor progression via the differentiation and motility of PSCs or resident fibroblasts that already exist in the pancreatic tissue [56]. However, despite the positive effects of an antibody against SHH in the PDAC preclinical animal model [56], a clinical trial showed that the SHH inhibitor did not synergize PDAC patients with gemcitabine treatment [57].
Bone marrow-derived mesenchymal stem cells (MSCs) may act as a potential source of CAFs in inflammation-induced GC [58]. The Helicobacter-induced GC mouse model reveals that CAFs are derived from α-SMA-positive myofibroblasts in the bone marrow, and these CAFs can form a tumor niche in the gastric wall and undergo tumor progression. The MSCs recruited from the bone marrow may act as a source of CAFs in PDAC and pancreatic endocrine tumors [54][59]. MSCs exposed to PDAC cells are activated into CAF-secreting tumor-promoting proteins such as hepatocyte growth factor (HGF), epidermal growth factor (EGF), and interleukin-6 (IL-6). These proteins stimulate microvascularization, changes in the composition of the stromal framework, and tumor growth through the paracrine system [54].
Other noncancerous cells, such as endothelial cells, pericytes, and even epithelial cells, which accumulate in GI cancer, can be transformed into CAFs through cell transition mechanisms. A study using a pancreatic islet tumor mouse model revealed that fibroblast-specific protein 1 (FSP1) and CD31 double-positive cells exist in the TME. Previous studies have reported that TGF-β mediates the transition from endothelial cells to mesenchymal cells in cardiac tissues [60]. Since abundant TGF-β expression was also apparent in this tumor, the authors suggested that TGF-β-exposed pancreatic endothelial cells could be a source of CAFs [61].
Vascular pericytes are multifunctional mural cells that surround endothelial cells [62], and they are crucial in the neoangiogenesis and survival of endothelial cells during tumorigenesis [63]. Emerging evidence has indicated that neural/glial antigen 2 (NG2)-expressing pericytes are transformed into CAFs through platelet-derived growth factor-BB (PDGF-BB) stimulation in a CRC xenograft model [64]. Moreover, the expression of PDGFB and FSP1 in various types of solid tumors, including CRC, is significantly correlated with poor patient prognosis [64].
Furthermore, epithelial cells of GI organs could be a source of CAFs during carcinogenesis. In genetic PDAC mouse models, pancreatic epithelial cells are transformed into mesenchymal cells through epithelial–mesenchymal transition; these cells have a fibroblast-like phenotype, express FSP1, and are deeply involved in tumor formation. However, although these FSP1-expressing cells are similar to CAFs, it is still unclear whether these cells could be a significant source of CAFs in PDAC tumors [65]. Therefore, further studies are required to verify whether epithelial cells are a crucial source of CAFs in GI cancers.
4. Factors Related to CAF-induced Resistance to Cancer Treatment
4.1. Cytokines and Chemokines
Cytokines and chemokines are inflammatory mediators secreted by cancer cells or tumor stromal cells in the TME. They can stimulate tumor-promoting pathways, including proliferation, metastasis, and progression in an autocrine or paracrine manner [66]. Moreover, the cytokines and chemokines in the TME are deeply related to chemoresistance and poor prognosis in cancer patients [67][68]. The CAFs in GI cancers could act as a source of various TME cytokines and chemokines.
IL-6, a multifaceted cytokine related to infection or injury response, plays a predominant role in cancer progression [69]. Recent studies have suggested that IL-6 is mainly secreted by CAFs, and CAF-derived IL-6 can induce an inadequate response to chemotherapy in GI cancers, including CRC, esophageal cancer (ESOC), and GC [15][70][71][72]. Qiao et al. [70] demonstrated that IL-6 from CAFs contributes to chemoresistance by activating the signal transducer and activator of transcription 3 (STAT3)/nuclear factor-κB (NF-κB) pathway and subsequently upregulating C-X-C motif chemokine receptor (CXCR) 7 expression in ESOC cells. Additionally, other studies demonstrated that stromal IL-6 increases the expression of cancer stem cell (CSC) markers and consequently induces resistance to chemoradiotherapy in ESOC patients [72]. Moreover, we demonstrated that CAF-derived IL-6 stimulates the Janus kinase 1 (JAK1)/STAT3 pathway in GC cells in a paracrine manner [15]. Furthermore, in human GC tissues, high expression of stroma-related genes, including IL-6, is significantly correlated with resistance to chemotherapy. Eventually, we found that the IL-6 receptor monoclonal antibody, tocilizumab, rescued CAF-induced resistance to chemotherapy in various experimental models [15]. Taken together, these studies show that CAFs may act as a source of IL-6 in the GI cancer microenvironment; as such, IL-6 inhibition could be a novel therapeutic strategy to decrease CAF-induced resistance to cancer therapies.
The human chemokine CXCL1, termed the GRO-1 oncogene, specifically binds to CXCR2, a member of the G-protein-coupled receptor family [73]. Zhang et al. [30] reported that the expression of CXCL1 in CAFs isolated from ESOC tissues was higher than that in normal fibroblasts. CAF-derived CXCL1 could be involved in tumor radiotherapy resistance by activating the MEK/ERK pathway.
CXCL12, also known as SDF-1, is mainly secreted from the stromal cells of solid tumors and is a primary ligand of the membrane receptor CXCR4 [74]. The role of CXCL12 has been explored in PDAC; several studies have reported that PSCs secrete CXCL12 into the TME, which promotes the resistance of PDAC cells to chemotherapy in a paracrine manner [75][76][77]. Secreted CXCL12 may activate the FAK/ERK1/2/AKT signaling pathways in PDAC cells, thereby inducing resistance to gemcitabine [75][77]. CXCL12-induced activation of this signaling pathway increases the transcriptional activities of β-catenin and NF-κB, thus leading to an elevated expression of survival proteins such as Bcl-2 [77]. Moreover, these CXCL12-activated pathways increased the secretion of IL-6 in PDAC cells related to chemoresistance [75]. Therefore, the small-molecule CXCR4 antagonist plerixafor has been used to abolish CXCL12-induced PDAC growth and chemoresistance [75][77]. A recent clinical trial demonstrated that the combination of plerixafor and chemotherapy increased the response rate of conventional chemotherapy in a hematological malignancy [78]; hence, this combination may be an effective chemosensitizer for GI cancer. Radiotherapy has been perioperatively administered to patients with PDAC. PDAC patients who undergo curative resection can be treated with radiotherapy to suppress cancer recurrence. Radiotherapy for inoperable PDAC patients can be used for symptom palliation [79]. One study concluded that CAF-derived CXCL12 promotes PDAC cell resistance to radiotherapy through CXCR4 activation [80]. This result suggests that CAF-induced CXCL12/CXCR4 signaling could be a novel therapeutic target to improve the effectiveness of radiation [80].
Chemotherapeutic agents can stimulate the production of various secretory proteins in CAFs. In experimental models of CRC, chemotherapy-stimulated CAFs enhance the secretion of specific cytokines such as IL-17A, and increased serum levels of IL-17 have been observed in CRC patients with chemoresistance. CAF-secreted IL-17A promotes chemoresistance in cancer-initiating cells (CICs) through the NF-κB pathway and increases CIC self-renewal, invasion, and tumor growth in vivo [81].
4.2. Growth Factors
Cancer cells usually express various receptor tyrosine kinases (RTKs) that can mediate downstream signaling pathways, such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-OH kinase, which can contribute to therapy resistance [82][83]. Although RTKs are highly activated through genetic mutations in various cancers, growth factor stimulation is a crucial mechanism for RTK-induced inadequate therapeutic responses [84]. In particular, if growth factors are secreted from CAFs, they can act as messengers for cell–cell communication.
In addition, cancer-secreted TGF-β can enhance the transition of resident fibroblasts into CAFs, as mentioned in Section 3, and CAF-secreted TGF-β is involved in cancer therapy resistance in GI cancer cells. In ESOC, CAF-conditioned media includes a higher concentration of TGF-β1 than the conditioned media from normal fibroblasts [31]. Consequently, CAF-derived TGF-β1 enhances resistance to cisplatin and taxol, and TGF-β1 expression in CAFs is significantly related to poor prognosis in ESOC patients subjected to chemoradiotherapy [31]. Another study showed that miR-27a/b converts normal fibroblasts to CAFs in ESOC, and the converted CAFs enhance resistance to cisplatin by secreting TGF-β1 [85]. Both studies demonstrated that the TGF-β1 inhibitor LY2157299 could improve the response of ESOC cells to various chemotherapeutic agents both in vivo and in vitro.
The insulin-like growth factor (IGF) family plays a crucial role in regulating cell proliferation and apoptosis by activating transmembrane receptors; thus, it contributes to resistance to GI cancer therapies [86]. Ireland et al. [87] suggested that CAFs could be a source of IGF-1 and IGF-2 in PDAC and consequently activate insulin/IGF receptors on PDAC cells. They also demonstrated that the inhibition of IGFs sensitizes PDAC cells to gemcitabine. The mechanism underlying the upregulation of IGF-1 expression in PDAC CAFs was evaluated by Xiao et al., [88] who demonstrated that the PDAC-enhanced methylation of suppressor of cytokine signaling 1 (SOCS1) plays a pivotal role in the transition of normal fibroblasts to CAFs. In turn, SOCS1 downregulation was associated with IGF-1 expression in CAFs. Moreover, radiotherapy may trigger the secretion of IGF1, which could be involved in the resistance of rectal cancer to radiotherapy. Radiation-activated CAFs promote CRC cell survival by activating the IGF-1 receptor; thereafter, the neutralization of this receptor in a CRC cancer animal model reduces metastasis [89].
HGF is a major secretory protein of CAFs in solid tumors that promotes cancer cell survival and provides therapeutic resistance [90]. CAF-secreted HGF increases the proportion of tumor-initiating cells of HCC through c-MET activation. Activated c-MET in tumor-initiating cells further activates the ERK/FRA1/HEY1 cascade, which is related to chemotherapy resistance [91].
Cetuximab, an EGF receptor (EGFR) monoclonal antibody, is a molecular targeted agent that improves the survival of patients with CRC without Kras mutation [92]. Luraghi et al. [93] reported that CAF-secreted HGF activates resistance to EGFR inhibitors in experimental models. CAF-induced HGF could also play a pivotal role in radiotherapy resistance. One study demonstrated that the levels of secreted HGF in irradiated fibroblasts isolated from ESOC were higher than those in non-irradiated controls. HGF derived from irradiated fibroblasts increases wound healing, migration, and invasion [94].
4.3. Exosomes
Various studies have examined the role of exosomes in cancer progression. The exosome, a microvesicle of endocytic origin with a diameter of 30–150 nm, is secreted by many cells. As exosomes comprise a lipid bilayer containing various bioactive molecules, such as DNA, microRNAs (miRNAs), proteins, long non-coding RNAs (lncRNAs), circular RNAs, and lipids, they function as natural vehicles in cell–cell communication by transferring genetic messages. Exosomes secreted from various cells within tumors enable communication among tumor cells surrounding the TME and in distant organs or tissues, leading to the promotion of metastasis and therapy resistance [95]. Therefore, exosomes generated from CAFs would be suitable messengers to enhance the resistance of GI cancer cells to therapy.
The function of CAF-derived exosomes in cancer therapy resistance was initially investigated in CRC. Hu et al. [96] reported that CAF-derived exosomes promote drug resistance by mediating the activation of the Wnt signaling pathway in CSCs in CRC. Next, the kinds of elements included in CAF-derived exosomes that can enhance resistance to therapies have recently been evaluated. Non-coding RNAs (ncRNAs) such as miRNAs and lncRNAs have been suggested as crucial molecules for exosome-mediated communication between CAFs and GI cancer cells [97]. miRNAs are a subtype of ncRNAs that contain 17–25 bp of ncRNA and usually suppress messenger RNA translation by targeting the 3′-UTR; thus, miRNAs facilitate the epigenetic regulation of gene expression and can control the pathological process in cancers [98]. Notably, several miRNAs present in exosomes are involved in drug resistance in cancers [99]. Regarding GI cancers, CAFs of CRC secrete miR-92a-3p-enriched exosomes into the TME. When exosomal miR-92a-3p is transferred to CRC cells, it promotes migration, invasion, metastasis, stemness, and drug resistance. Thus, blocking the function of exosomal miR-92a-3p secreted by CAFs could be used as an alternative modality for therapy resistance in CRC [100]. In GC, Zhang et al. [101] demonstrated that CAF-secreted exosomal miR-522 regulates arachidonate lipoxygenase 15 (ALOX15) expression and is closely related to lipid reactive oxygen species (ROS) production. They suggested that blocking lipid-ROS production might be a novel mechanism for acquired drug resistance. Thus, targeting exosomal miR-522 could be a modality to increase the sensitivity of GC patients to chemotherapy [101]. Furthermore, when miR-106b in the PDAC CAF-derived exosomes is transferred into PDAC cells, it mediates resistance to gemcitabine [102]. Therefore, detecting miRNAs in CAF-derived exosomes of GI cancer could provide efficient biomarkers to predict chemotherapy response. Moreover, targeting the function of these miRNAs could serve as a promising tool for improving the drug response in GI cancers.
Exosomes can also contain lncRNAs, which are nucleotide transcripts over 200 bp in length that are not translated into proteins [103]. The function of lncRNAs in the resistance of various cancers to therapies has recently been proposed; this could be the central mechanism related to CAF exosome-derived drug resistance in GI cancer. Deng et al. [104] demonstrated that CAF-derived exosomes express CRC-associated lncRNA (CCAL) more highly than normal fibroblasts, and these CCAL-enriched exosomes may drive cancer cells to oxaliplatin resistance. In addition, the transfer of CCAL could function as an oncogenic lncRNA and induce Wnt/β-catenin pathway activation in CRC cells. Therefore, CCAL may represent a biomarker and druggable target for CRC chemoresistance. Another CAF-derived exosomal lncRNA, H19, also promotes stemness and chemoresistance in CRC cells [105]. Transferred H19 could activate the β-catenin pathway in CRC cells by blocking the function of miR-141, which could inhibit stemness.
Furthermore, CAF-derived exosomes may also be associated with resistance to radiotherapy. Liu et al. [106] reported that CAF-derived exosomes confer robust radiation resistance in CRC cells by activating the TGF-β signaling pathway.
4.4. Other Mechanisms
The other secretory materials produced from CAFs and CAF-induced intratumoral pressure escalation could be involved in the resistance to GI cancer therapies.
Plasminogen activator inhibitor-1 (PAI-1) is a secreted protein that not only enhances angiogenesis, but also promotes the invasion and metastasis of certain cancer cells [107][108]. PAI-1 is secreted from CAFs, and one study reported that cisplatin-treated CAFs increase PAI-1 secretion [32]. CAF-secreted PAI-1 enhances progression and chemoresistance by activating the AKT/ERK1/2 signaling pathway and inhibiting caspase-3 activity and ROS accumulation in ESOC cells. Moreover, the high expression of PAI-1 in CAFs is correlated with poor prognosis in ESOC patients; consistently, the PAI-1 inhibitor tiplaxtinin presents synergistic effects with cisplatin both in vitro and in vivo [32].
CAF-secreted perlecan (heparin sulfate proteoglycan 2, HSPG2) plays a critical role in resistance to chemotherapy in PDAC. CAFs isolated from genetic PDAC mouse models were reprogrammed in mouse PDAC cells with a P53 mutation. The reprogrammed CAFs increased the stromal deposition of HSPG2, which created a prometastatic and chemoresistant environment in pancreatic cancer cells [109]. Another CAF-induced secreted protein that protects PDAC cells from gemcitabine is laminin A1. Although PDAC cells secrete transglutaminase, they do not increase the cytotoxicity of gemcitabine directly; however, transglutaminase enhances the secretion of laminin A1 from CAFs, and secreted laminin A1 secreted in the TME could protect PDAC cells from chemotherapeutic agents such as gemcitabine [110].
CAF-secreted T-lymphoma invasion and metastasis-inducing protein-1 (TIAM1) is a key regulator of chemoresistance in CRC cells [111]. CAF-derived conditioned media increased resistance to chemotherapy through TIAM1 overexpression, and TIAM1-associated drug sensitivity was validated using a xenograft mouse model.
CAFs are the center of desmoplastic reactions in GI cancer as well as a source of secreted proteins; thus, they interfere with drug delivery by collapsing the peritumoral capillaries and increasing intratumoral interstitial pressure. This concept has been suitably evaluated in a mouse model of PDAC, wherein GI tumors exhibit profuse desmoplastic reactions. The accumulation of hyaluronic acid (HA) produced from CAFs during PDAC progression was found to be responsible for enhancing intratumoral pressure, thus acting as a barrier for drug diffusion in the mouse model. Provenzano et al. [112] suggested that enzymatic dissolution of stromal HA could increase the efficacy of cancer drugs by remodeling PDAC stromal lesions.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Global Burden of Disease Cancer, C.; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568.
- Myint, A.S. The role of radiotherapy in the palliative treatment of gastrointestinal cancer. Eur. J. Gastroenterol Hepatol. 2000, 12, 381–390.
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. S1), 35–48.
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792.
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586.
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat Oncol. 2020, 22, 90–97.
- Ribeiro Franco, P.I.; Rodrigues, A.P.; de Menezes, L.B.; Pacheco Miguel, M. Tumor microenvironment components: Allies of cancer progression. Pathol. Res. Pract. 2020, 216, 152729.
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566.
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93.
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186.
- Melissari, M.T.; Chalkidi, N.; Sarris, M.E.; Koliaraki, V. Fibroblast Reprogramming in Gastrointestinal Cancer. Front. Cell Dev. Biol. 2020, 8, 630.
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601.
- Mueller, M.M.; Fusenig, N.E. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849.
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68.
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753.
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401.
- Liu, H.; Ma, Q.; Xu, Q.; Lei, J.; Li, X.; Wang, Z.; Wu, E. Therapeutic potential of perineural invasion, hypoxia and desmoplasia in pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2395–2403.
- Vennin, C.; Murphy, K.J.; Morton, J.P.; Cox, T.R.; Pajic, M.; Timpson, P. Reshaping the Tumor Stroma for Treatment of Pancreatic Cancer. Gastroenterology 2018, 154, 820–838.
- Ueno, H.; Jones, A.M.; Wilkinson, K.H.; Jass, J.R.; Talbot, I.C. Histological categorisation of fibrotic cancer stroma in advanced rectal cancer. Gut 2004, 53, 581–586.
- Sis, B.; Sarioglu, S.; Sokmen, S.; Sakar, M.; Kupelioglu, A.; Fuzun, M. Desmoplasia measured by computer assisted image analysis: An independent prognostic marker in colorectal carcinoma. J. Clin. Pathol. 2005, 58, 32–38.
- Hofheinz, R.D.; Al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48.
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027.
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366.
- Ma, J.; Song, X.; Xu, X.; Mou, Y. Cancer-Associated Fibroblasts Promote the Chemo-resistance in Gastric Cancer through Secreting IL-11 Targeting JAK/STAT3/Bcl2 Pathway. Cancer Res. Treat. 2019, 51, 194–210.
- Gu, J.; Li, Z.; Zhou, J.; Sun, Z.; Bai, C. Response prediction to oxaliplatin plus 5-fluorouracil chemotherapy in patients with colorectal cancer using a four-protein immunohistochemical model. Oncol. Lett. 2019, 18, 2091–2101.
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Yokoe, T.; Okugawa, Y.; Kawamoto, A.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Stromal CXCR4 and CXCL12 expression is associated with distant recurrence and poor prognosis in rectal cancer after chemoradiotherapy. Ann. Surg. Oncol. 2010, 17, 2051–2058.
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Yokoe, T.; Okugawa, Y.; Fujikawa, H.; Matsusita, K.; Kawamura, M.; Inoue, Y.; Miki, C.; et al. Cancer-associated fibroblasts correlate with poor prognosis in rectal cancer after chemoradiotherapy. Int. J. Oncol. 2011, 38, 655–663.
- Luo, Y.; Mao, Q.; Wang, X.; Yu, J.; Li, M. Radiotherapy for esophageal carcinoma: Dose, response and survival. Cancer Manag. Res. 2018, 10, 13–21.
- Zhang, H.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Xu, Y.; Wu, S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017, 8, e2790.
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFbeta1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog 2017, 56, 1150–1163.
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018, 9, 759.
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8.
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175.
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612.
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782.
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017, 6.
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218.
- Wu, Y.; Grabsch, H.; Ivanova, T.; Tan, I.B.; Murray, J.; Ooi, C.H.; Wright, A.I.; West, N.P.; Hutchins, G.G.; Wu, J.; et al. Comprehensive genomic meta-analysis identifies intra-tumoural stroma as a predictor of survival in patients with gastric cancer. Gut 2013, 62, 1100–1111.
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356.
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329.
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178.
- Ji, J.; Eggert, T.; Budhu, A.; Forgues, M.; Takai, A.; Dang, H.; Ye, Q.; Lee, J.S.; Kim, J.H.; Greten, T.F.; et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015, 62, 481–495.
- Li, Z.; Gao, X.; Peng, X.; May Chen, M.J.; Li, Z.; Wei, B.; Wen, X.; Wei, B.; Dong, Y.; Bu, Z.; et al. Multi-omics characterization of molecular features of gastric cancer correlated with response to neoadjuvant chemotherapy. Sci. Adv. 2020, 6, eaay4211.
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86.
- Kadel, D.; Zhang, Y.; Sun, H.R.; Zhao, Y.; Dong, Q.Z.; Qin, L.X. Current perspectives of cancer-associated fibroblast in therapeutic resistance: Potential mechanism and future strategy. Cell Biol. Toxicol. 2019, 35, 407–421.
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256.
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018.
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Kashiwagi, S.; Matsuoka, J.; Doi, Y.; Kato, Y.; Hasegawa, T.; Sawada, T.; Hirakawa, K. Upregulation of cancer-associated myofibroblasts by TGF-beta from scirrhous gastric carcinoma cells. Br. J. Cancer 2011, 105, 996–1001.
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992.
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541.
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004.
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292.
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272.
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495.
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128.
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Pericytes at the intersection between tissue regeneration and pathology. Clin. Sci. 2015, 128, 81–93.
- von Tell, D.; Armulik, A.; Betsholtz, C. Pericytes and vascular stability. Exp. Cell Res. 2006, 312, 623–629.
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627.
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361.
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185.
- Reyes, M.E.; de La Fuente, M.; Hermoso, M.; Ili, C.G.; Brebi, P. Role of CC Chemokines Subfamily in the Platinum Drugs Resistance Promotion in Cancer. Front. Immunol. 2020, 11, 901.
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265.
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572.
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883.
- Huynh, P.T.; Beswick, E.J.; Coronado, Y.A.; Johnson, P.; O’Connell, M.R.; Watts, T.; Singh, P.; Qiu, S.; Morris, K.; Powell, D.W.; et al. CD90(+) stromal cells are the major source of IL-6, which supports cancer stem-like cells and inflammation in colorectal cancer. Int. J. Cancer 2016, 138, 1971–1981.
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242.
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157.
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110.
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097.
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging roles of the CXCL12/CXCR4 axis in pancreatic cancer progression and therapy. Pharmacol. Ther. 2017, 179, 158–170.
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679.
- Ghobrial, I.M.; Liu, C.J.; Zavidij, O.; Azab, A.K.; Baz, R.; Laubach, J.P.; Mishima, Y.; Armand, P.; Munshi, N.C.; Basile, F.; et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am. J. Hematol. 2019, 94, 1244–1253.
- Venkatesulu, B.P.; Hsieh, C.E.; Sanders, K.L.; Krishnan, S. Recent advances in radiation therapy of pancreatic cancer. F1000Research 2018, 7.
- Li, D.; Qu, C.; Ning, Z.; Wang, H.; Zang, K.; Zhuang, L.; Chen, L.; Wang, P.; Meng, Z. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am. J. Cancer Res. 2016, 6, 2192–2206.
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872.
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010, 3, ra64.
- Engelman, J.A.; Settleman, J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr. Opin. Genet. Dev. 2008, 18, 73–79.
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784.
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemura, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903.
- LeRoith, D.; Baserga, R.; Helman, L.; Roberts, C.T., Jr. Insulin-like growth factors and cancer. Ann. Intern. Med. 1995, 122, 54–59.
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863.
- Xiao, Q.; Zhou, D.; Rucki, A.A.; Williams, J.; Zhou, J.; Mo, G.; Murphy, A.; Fujiwara, K.; Kleponis, J.; Salman, B.; et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016, 76, 5395–5404.
- Tommelein, J.; De Vlieghere, E.; Verset, L.; Melsens, E.; Leenders, J.; Descamps, B.; Debucquoy, A.; Vanhove, C.; Pauwels, P.; Gespach, C.P.; et al. Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res. 2018, 78, 659–670.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189.
- Chan, D.L.H.; Segelov, E.; Wong, R.S.; Smith, A.; Herbertson, R.A.; Li, B.T.; Tebbutt, N.; Price, T.; Pavlakis, N. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database Syst. Rev. 2017, 6, CD007047.
- Luraghi, P.; Reato, G.; Cipriano, E.; Sassi, F.; Orzan, F.; Bigatto, V.; De Bacco, F.; Menietti, E.; Han, M.; Rideout, W.M., 3rd; et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014, 74, 1857–1869.
- Patel, Z.S.; Grugan, K.D.; Rustgi, A.K.; Cucinotta, F.A.; Huff, J.L. Ionizing radiation enhances esophageal epithelial cell migration and invasion through a paracrine mechanism involving stromal-derived hepatocyte growth factor. Radiat Res. 2012, 177, 200–208.
- Kahlert, C.; Kalluri, R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J. Mol. Med. 2013, 91, 431–437.
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625.
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37.
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333.
- Bach, D.H.; Hong, J.Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230.
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91.
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43.
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543.
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251.
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716.
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H. Theranostics 2018, 8, 3932–3948.
- Liu, L.; Zhang, Z.; Zhou, L.; Hu, L.; Yin, C.; Qing, D.; Huang, S.; Cai, X.; Chen, Y. Cancer associated fibroblasts-derived exosomes contribute to radioresistance through promoting colorectal cancer stem cells phenotype. Exp. Cell Res. 2020, 391, 111956.
- Hirahata, M.; Osaki, M.; Kanda, Y.; Sugimoto, Y.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Kawai, A.; Ito, H.; et al. PAI-1, a target gene of miR-143, regulates invasion and metastasis by upregulating MMP-13 expression of human osteosarcoma. Cancer Med. 2016, 5, 892–902.
- Geis, T.; Doring, C.; Popp, R.; Grossmann, N.; Fleming, I.; Hansmann, M.L.; Dehne, N.; Brune, B. HIF-2alpha-dependent PAI-1 induction contributes to angiogenesis in hepatocellular carcinoma. Exp. Cell Res. 2015, 331, 46–57.
- Vennin, C.; Melenec, P.; Rouet, R.; Nobis, M.; Cazet, A.S.; Murphy, K.J.; Herrmann, D.; Reed, D.A.; Lucas, M.C.; Warren, S.C.; et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 2019, 10, 3637.
- Lee, J.; Yakubov, B.; Ivan, C.; Jones, D.R.; Caperell-Grant, A.; Fishel, M.; Cardenas, H.; Matei, D. Tissue Transglutaminase Activates Cancer-Associated Fibroblasts and Contributes to Gemcitabine Resistance in Pancreatic Cancer. Neoplasia 2016, 18, 689–698.
- Izumi, D.; Toden, S.; Ureta, E.; Ishimoto, T.; Baba, H.; Goel, A. TIAM1 promotes chemoresistance and tumor invasiveness in colorectal cancer. Cell Death Dis. 2019, 10, 267.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429.

