 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nipa H. Patel | + 10999 word(s) | 10999 | 2020-11-30 08:41:37 | | | |
| 2 | Rita Xu | -5604 word(s) | 5395 | 2020-12-15 07:29:35 | | |
Video Upload Options
Multidrug resistance is a major factor contributing to the failure of cancer therapy and poor patient outcomes. While apoptosis (apoptotic cell death) is the desired outcome of anti-cancer therapy, chemotherapy and radiation often induce a number of mechanisms that can mediate resistance. p53 is an essential tumor suppressor and stress response protein, modulating multiple cellular responses to therapy. Gain of function (GOF) p53 mutations have been implicated in increased susceptibility to the development of drug resistance, by compromising wild type anti-tumor functions of p53 or modulating key p53 processes that confer chemotherapy resistance, such as autophagy. Autophagy, a conventionally cytoprotective mechanism, is often a “first responder” to chemotherapy (or radiation), by promoting the removal of damaged organelles and preventing excessive accumulation of damaged proteins; thus, autophagy, via its cytoprotectivefunction, may allow tumor cells to evade apoptotic cell death. However, substantial pre-clinical data and inconsistent clinical efficacy of autophagy inhibitors in combination with cancer chemotherapy indicates that autophagy can exhibit multiple functions and does not act solely as a cytoprotective response. Further inquiry relating to the influence of p53 status on autophagic function and its contributions to multidrug resistance will provide valuable insights towards patient response to therapy and the possibility of developing novel therapeutics for chemosensitization in the face of multidrug resistance.
1. Introduction
Although the treatment of cancer has seen significant advances in recent years, chemotherapeutic drugs continue to represent a primary component of most current cancer therapies. However, drug resistance, and often multidrug resistance (MDR), are primary reasons for the failure of clinical chemotherapy [1].
Drug resistance can be intrinsic to the tumors or be acquired during treatment. Long-term sublethal drug exposure of tumor cells is one basis for the development of acquired drug resistance. Drug resistance often involves multiple mechanisms. For instance, physiological barriers such as a dense fibroblast envelope in the tumor tissue and the absence of lymphatic drainage can limit drug access to the tumor [2]. This is particularly relevant to efforts to treat pancreatic cancer [3][4]. The generation of acidic lysosomes in drug-resistant tumor cells forming ion traps that can bind weakly basic anticancer drugs (such as adriamycin hydrochloride and irinotecan hydrochloride) may contribute to reduced drug efficacy [5]. In addition, activation of multidrug-resistant proteins, inhibition of cell death pathways through imbalance in pro-apoptotic and anti-apoptotic proteins, changes in drug metabolism, epigenetic changes, or changes in drug targets may all lead to chemotherapy resistance.
In recent years, extensive evidence has accumulated for senescence as a primary response to cancer therapeutics. Therapy-induced senescence (TIS) provides an additional layer of complexity to predicting tumor cell fate after therapy in that recovery from senescence provides a potential pathway from drug and radiation lethality. Since subpopulations of tumor cells can enter into a transient senescent-mediated growth arrested state and subsequently regain proliferative capacity, senescence may represent one element of tumor dormancy state, which eventually facilitates disease recurrence, representing a mechanism of delayed drug resistance [6].
The primary intent of cancer treatment strategies is obviously to promote tumor cell death; therefore, factors affecting cell death and the underlying mechanisms are central issues in determining therapeutic efficacy. As the most well-studied tumor suppressor gene, p53 plays a “gate keeper” role in tumorigenesis [7][8]. Unfortunately, the p53 pathway is often inactivated or functions in an antagonistic manner towards drug effectiveness [9][10]. In fact, p53 was initially identified as an oncogene due to the inadvertent use of mutated p53 sequences from tumors [11]. The mutated form of p53 protein is often expressed in cancer, promoting cell transformation, metastasis and drug resistance, in part by inhibiting wildtype p53 (wtp53) [12]. Cancer genome sequencing has shown that 42% of the 12 tumor types studied carried the mutant TP53 gene [9][13].
In tumor cells exposed to chemotherapy and radiotherapy, one of the first responses is autophagy, a cellular process that removes damaged proteins and organelles as well as generating energy and metabolic intermediates. Autophagy can promote or attenuate tumor resistance, depending on whether it is cytoprotective or cytotoxic in nature [14][15]. There is extensive evidence that p53 can modulate autophagy. Interestingly, p53 can play dual roles, where nuclear p53 induces autophagy through transcriptional effects, whereas cytoplasmic p53 acts as a master repressor of autophagy [16][17]. Hence, it is not surprising that tumors differing in p53 status may not have identical influence on autophagy function. This review attempts to provide a comprehensive summary of the effects of p53 status on the functional form of autophagy, which in turn modulates drug sensitivity and resistance.
1.1. p53 and Drug Resistance
The p53 tumor suppressor protein, a transcription factor that can respond to various forms of exogenous stress and inhibit cell division or survival, is often considered to be the key fail-safe mechanism of cell anti-cancer defenses [18][19]. Consequently, in order to enhance their survival and/or maintain growth, cancer cells use a variety of strategies to disarm p53. The most direct and effective way to inactivate p53 is to mutate the p53-encoding gene TP53 [20]. Since the frequent mutation of TP53 in human cancers was described 30 years ago, the mutation patterns of TP53 in cancers and the role of p53 in cancer etiology have been gradually clarified [21][22][23]. Mutations in p53 are the most common genetic lesion in cancers, and correspond with cancer development, progression, metastasis, and resistance to chemotherapy or radiotherapy. Most p53 mutations occur in the central DNA-binding domain, resulting in the loss of wildtype function (so-called loss of function, LOF) or have a dominant-negative effect on the wildtype alleles. Some mutations (such as R248Q, R273H, R175H, and R249S) have shown “gain of function” (GOF), which can further promote cancer malignancy and chemoresistance [10]. However, there are several DNA binding domain mutants, such as G245S and R246S variants, which do not exhibit any GOF properties [24][25]. The reason why only some p53 mutants express GOF properties are still unclear, but we might anticipate that drugs that directly target mutant p53 for degradation might be useful in improving the therapeutic responses.
The multidrug resistance gene 1 (MDR1), also known as ABCB1, is often found to be over-expressed in cancer, encoding an ATP-dependent efflux pump, which is responsible for inducing broad-spectrum chemical resistance. After Chin et al. demonstrated transcriptional dependence of the MDR1 gene promoter on p53, the clinical correlations between therapeutic resistance and p53 mutations gained more prominence and attention [26][27]. It has also been shown that p53 mutants are able to actuate various survival signaling cascades, such as the NF-κB, PDGFRβ, mevalonate, proteasomal, or integrin pathways [28][29][30][31], and activate an independent set of target genes in cooperation with other transcription factors or cofactors (such as Pin1 [32] and PML [33] proteins), thereby promoting tumor cell survival and/or proliferation.
The mutant form of p53 can confer resistance to apoptosis, thereby reducing tumor cell susceptibility to cell death [27][34]. Mutant p53 not only interferes with the transcriptional activity of wtp53 in the nucleus, but also abolishes the interaction between wtp53 and BCL-2 family proteins in the cytoplasm. For example, p53 dysfunction led to decreased apoptosis induction by BCL-2 antagonists ABT-737 in chronic lymphocytic leukemia cells [35]. The p53 mutation of GOF also mediates resistance to apoptosis for many commonly used chemotherapeutic agents. For instance, cross-resistance between doxorubicin and paclitaxel was induced by introduction of the R248Q p53 mutant into hepatocellular carcinoma p53-null Hep3B cells [36]. Another example is where knockdown of the R273H p53 mutant in human squamous cell carcinoma increased procaspase-3 levels and sensitized these cells to doxorubicin and methotrexate-induced apoptosis [37]. Due to the LOF or GOF of abnormal TP53 in tumors, reintroducing p53 through a virus encoding wtp53 or converting mutant p53 to wildtype function may be a potential therapeutic strategy for increasing the susceptibility of tumor cells to apoptosis [38]. However, regardless of the attempts to restore p53 in tumors lacking p53 [39] with p53 missense mutations [40] or in tumors driven by oncogenes [41][42], it is still difficult to predict the nature of the p53-mediated response that will be evoked, whether it is conventional growth arrest, senescence, and/or apoptosis. It seems the most effective approach might be to combine the reintroduction of p53 function with conventional chemotherapy drugs to promote tumor cell apoptosis. Taken together, in chemotherapy, mutant p53 represents a key factor in cancer cell resistance to treatment.
1.2. Autophagy and p53 in Cancer Treatment
Autophagy, a process of self-degradation, represents a critical physiological catabolic mechanism of eukaryotic cells. Autophagy is necessary for cells to respond to nutrient starvation and other types of stressful conditions, such as hypoxia [43]. Consequently, it is not surprising that autophagy can often be detected in tumor cells exposed to chemotherapy or radiation [44]. In response to chemotherapy, autophagy may exhibit several functional forms, including a cytoprotective form, a cytotoxic form that either directly or indirectly promotes tumor cell death, and what we have termed a nonprotective form, which does not appear to directly influence cell proliferation or apoptosis [15]. However, it is still unclear why autophagy, a life process that maintains cell homeostasis via elimination of oncogenic protein substrates, toxic unfolded proteins, and damaged organelles, exhibits these inconsistent effects. Nevertheless, because of the various roles played by autophagy in cancer treatment, autophagy offers a high degree of potential for future therapy.
Indeed, at present, the majority of clinical studies that involve autophagy are in the field of cancer therapy. Currently, the mainstream clinical trials involve the combined use of chemotherapeutic drugs and chloroquine (CQ) or hydroxychloroquine (HCQ), based on the largely unproven premise that all therapies promote the cytoprotective form of autophagy in patient malignancies [45]. Published clinical trials have demonstrated the safety of CQ or HCQ; moreover, increased radiosensitivity and prolonged patient survival has been evident primarily in the treatment of glioblastoma [46][47][48]. Generally, however, the results of these clinical trials of CQ or HCQ combined with chemotherapy have been largely inconsistent, indicating the challenge of extrapolating to the clinical situation from in vitro and in vivo preclinical studies. In part, the inconsistency evident in clinical trials may be attributed to the inability to achieve sufficient HCQ or CQ plasma levels required to inhibit autophagy [49]. In fact, significantly higher doses would likely be required to effectively achieve autophagy inhibition in patient tumors, which would result in severe toxic side effects [49]. Furthermore, the optimal time frames for administration of autophagy inhibitors to maximize sensitization to chemotherapy or radiation therapy have also generally not been considered in the design of these clinical trials. Studies have shown that for normal non-cancerous cells, autophagy is necessary for the maintenance of cellular homeostasis, while mice lacking ATG5 or ATG7 have spontaneous defects [50][51][52]. Shingu et al. found that inhibition of autophagy at a late stage enhanced imatinib-induced cytotoxicity in human malignant glioma cells, but attenuated the imatinib-induced cytotoxicity at an early stage [53]. These studies suggest that if autophagy inhibitors are applied in the early stage of tumor formation, there is a risk of promoting tumorigenesis.
Another issue that should not be ignored is the relationship between autophagy and the immune system. It has been demonstrated that autophagy has a surveillance effect on immunity, and a reduction of autophagy is related to the infiltration of regulatory T cells, which inhibit the immune system and reduce effective immune surveillance, thereby stimulating a tumor-promoting microenvironment [54]. Autophagy also plays a key role in processing of DAMPs, cytokine, and chemokine release for immune infiltration and recruitment, as well as processing of tumor-antigens for MHC presentation [55]. In addition, while autophagy deficiency has been shown to increase chemo- and radiosensitivity in immune-deficient mice [56][57], inhibition of autophagy in immune-competent mice has been reported to result in a failure of chemotherapy [58][59]; oddly, despite having been published almost 10 years ago, the latter study has not been independently verified by additional reports. This background further suggests that in the initial stage of tumor formation, when the immune system plays a critical role, inhibiting autophagy may accelerate the occurrence and progression of tumors. For now, the optimal staging for the use of autophagy inhibitors is in advanced tumors, which is also an ideal stage for the clinical trials that have been completed.
Although many factors are likely to influence autophagy, the involvement of p53 cannot be ignored. As mentioned above, p53 plays a regulatory role in tumor cell proliferation, cell cycle regulation, apoptosis, senescence, and autophagy. This regulatory role is related to its subcellular localization. Studies have shown that p53 located in the nucleus promotes autophagy under stress, while cytosolic p53 inhibits autophagy in unstressed cells [16][17]. In the nucleus, p53 induces autophagy by regulating the mTOR pathway in a transcription-dependent manner, as well as transcriptional regulation of key autophagy-related genes (ATGs) [39][60]. Some p53 targeted genes, including PTEN, TSC2, and AMPKβ, have been reported to negatively regulate mTOR [61], thus promoting autophagy initiation. In addition, p53 can also be associated with autophagy through the p14ARF (p19ARF in mouse, and hereafter referred to as ARF)-signaling pathway [62]. During tumorigenesis, the activation of oncogenes upregulates the transcription of ARF, which in turn binds to and inhibits the expression of MDM2, thereby stabilizing p53 [19]. ARF positively regulates autophagy by disrupting the Bcl-xl/Beclin 1 complex, releasing Beclin-1 to induce autophagy [62].
Subcellular localization of p53 can also mediate functional responses between apoptosis and autophagy. In the cytosol, p53 can localize to the mitochondria where it can interact with antiapoptotic BCL family proteins, allowing oligomerization of proapoptotic factors, such as BAX and BAK, thereby promoting mitochondrial outer membrane permeabilization (MOMP) and driving activation of intrinsic apoptotic cell death pathway [63][64]. Studies by Tomita et al. demonstrated several breast cancer cell lines expressing various p53 mutants failed to form complexes with Bcl2 in MDA-MB-231 (p53 R280K), MDA-MB-468 (p53 R273H), T47D (p53 L194F), and SKBr3 (p53 R175H) cells when compared to ML-1 (wtp53) cells [65]. Furthermore, these p53 mutant breast cancer cell lines exhibited impaired mitochondrial permeabilization when compared to wtp53 cells.
High mobility group box 1 (HMGB1), a conserved nuclear protein, acts as a chromatin-binding factor, binds to DNA, and promotes access to transcriptional protein complexes [66]. Similar to p53, the biological function of HMGB1 is related to its subcellular location [67]. Besides its nuclear effect, HMGB1 plays an important role in the processes of inflammation, cell differentiation, cell migration, wound healing, and tumor progression [68][69][70]. In addition to being a Beclin1-binding protein [71], cytoplasmic HMGB1 maintains the activation of the Beclin1-PtdIns3KC3 complex during the upregulation of autophagy [72]. HMGB1 also forms a complex with p53 and affects the cytoplasmic localization of reciprocal binding partners, thereby regulating the subsequent levels of autophagy and apoptosis [73]. Moreover, the target genes of p53 including DRAM, ISG20L1, or AEN are all reported to have the ability to regulate autophagy [74][75][76].
1.3. Autophagy and Multidrug Resistance (MDR)
The recurrence of tumors after treatment continues to represent a critical problem for clinicians. This is often due, in large part, to the multidrug resistance (MDR) response of tumor cells to chemotherapeutic agents. Therefore, there is an urgent need to develop agents with high activity against MDR, but with limited overall toxicity. The phenomena and mechanisms of MDR are summarized in great detail in many reviews [77][78][79], but in this review, we focused on how autophagy mediates MDR. Taken together with our discussion above, when autophagy exhibits cytoprotective functions, the administration of autophagy inhibitors can enhance chemotherapeutic drug sensitivity. In a Ras-NIH 3T3-Mdr cell model overexpressing p-glycoproteins (p-gp), a deficiency of autophagy was also found to facilitate necrosis and apoptosis induced by gossypol, a BH3-mimetic small molecule isolated from cottonseed, suggesting that autophagy may exhibit protective effects in drug-resistant cells [80]. Further studies have demonstrated that autophagy may be associated with resistance to a variety of anti-breast cancer drugs, such as tamoxifen, Herceptin (trastuzumab), paclitaxel (PTX), and epirubicin (EPI) [81]. These results were also validated in multidrug-resistant v-Ha-ras-transformed NIH 3T3 cell studies showing that knockout of the autophagy regulatory gene, ATG5, increased PTX sensitivity [82].
Conversely, when autophagy expresses a cytotoxic function, this could be exploited to induce MDR cancer cell death [83]. Sirichanchuen et al. found that co-treatment with cisplatin and autophagy inducer, trifluorperazine, could resensitize H460/cis cells to cisplatin-induced cell death [84]. This study is not unique, as Meschini et al. reported that vocamine, a bisindolic alkaloid from Peschiera fuchsiaefolia, could be utilized to overcome the resistance to doxorubicin in osteosarcoma cells by competitively inhibiting p-gp/ABCB1 and inducing autophagic cell death [85].
The mTOR pathway is a negative regulator of autophagy [86]. Rapamycin, an inhibitor of mTOR, activates autophagy and induces autophagic death of MDR v-Ha-ras-transformed NIH3T3 cells, drug-resistant LoVo/ADR colon cancer cells, and cisplatin-resistant cervical cancer cells [87][88][89]. GOF properties of p53 may also be involved in this process. The mutant p53 protein inhibits the generation of cytotoxic autophagy by stimulating the mTOR pathway, thereby increasing the proliferation of tumor cells [90]. In addition, when MDR cells lack the capacity to undergo apoptosis or exhibit apoptosis resistance, the agents that induce cytotoxic autophagy can also treat cancer and impede multidrug resistance through the induction of autophagy. Saikosaponin-d and Hernandezine are small molecular compounds extracted from natural plants, which promote the death of apoptosis-defective or apoptosis-resistant mouse embryonic fibroblast cells through cytotoxic autophagy [91][92].
Additional evidence linking autophagy to MDR suggests that lysosomal activity plays a role in tumor drug resistance. Lysosomes are at the center of cell degradative processes, responsible for decomposing proteins, polysaccharides, and lipids into their own basic structural forms [93]. Lysosomes receive extracellular or cell surface materials by endocytosis and intracellular components by autophagy. The unique acidic condition of lysosomes provides the optimal environment for the hydrolases in lysosome, and is also a precondition for the fusion of autophagosomes with lysosomes, forming the autophagolysosome and the completion of autophagic flux (i.e., degradation of the autolysosomal cargo) [94]. In addition to the widely accepted passive sequestration of hydrophobic weak base chemotherapeutics [95][96][97], other lysosome-mediated resistance mechanisms have been reported, such as the active lysosomal drug isolation mediated by the ATP-driven transporter of the ATP-binding cassette (ABC) superfamily [98][99] and the role of the lysosomal copper transporter in tumor resistance to platinum drugs [5][100][101]. Some genes regulated by p53 encode lysosomal proteins. In p53-deleted or mutant tumor cells, although autolysosome formation is not affected, a deficiency at the lysosome-mediated degradation of autophagosome cargo ensues [102]. Therefore, resistance induced by p53-dependent protective autophagy may be associated with the induction of high levels of lysosomes.
In general, both autophagy inhibitors and inducers are involved in the treatment of MDR-expressing cancer. There are many regulatory factors that affect the role of autophagy. Understanding the specific role of autophagy and taking advantage of autophagic functionality could potentially be an effective strategy to overcome multidrug resistance in cancer (Figure 1).
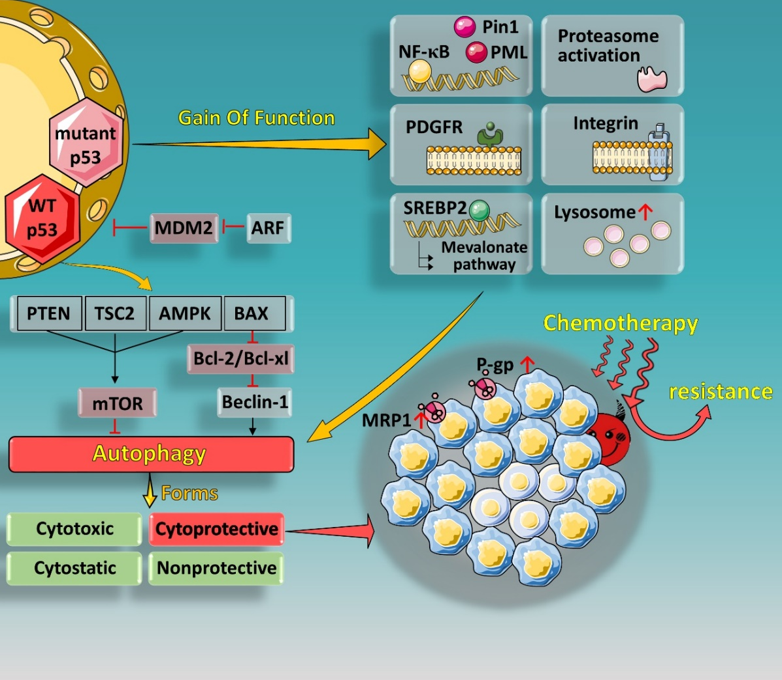
Figure 1. Gain of function (GOF) effect of mutant p53 and regulation of autophagy by p53 in nucleus. Certain p53 mutations (such as R248Q, R273H, R175H, and R249S) have shown GOF can further promote cancer malignance and chemoresistance. p53 mutants are able to actuate various survival signaling cascades, such as the NF-κB, PDGFRβ, mevalonate, proteasomal, or integrin pathways, and activate an independent set of target genes in cooperation with other transcription factors or cofactors (such as Pin1 and PML proteins). Nuclear p53 localization can promote autophagy by regulating the mTOR pathway in a transcription-dependent manner. Some p53 targeted genes, including PTEN, TSC2, and AMPKβ, have been reported to negatively regulate mTOR, thus promoting autophagy initiation. In addition, p53 can also be associated with autophagy through the ARF-signaling pathway. Regulation between p53 and the different functional forms of autophagy can contribute to chemotherapy resistance in tumor cells.
2. Effect of p53 Status on Autophagy and MDR
Sequencing of the cancer genome showed that 42% of the 12 tumor types analyzed carried the TP53 mutant gene [13]. However, the mutation rate of TP53 varies greatly among different types of tumors [9]. The importance of different types of p53 mutations leading to different therapeutic effects has been recognized; therefore, Food and Drug Adminstration (FDA)-approved drugs are now being tested in pre-clinical or clinical trials with patients stratified according to the status of p53 [10]. However, the p53 status of patients is rarely considered in clinical trials where autophagy inhibitors are used in combination. In subsequent sections, we will summarize the reported effects of p53 status on chemotherapeutic drug sensitivity and resistance in different types of tumors.
2.1. Leukemia
A wide range of studies have determined that about 10% of patients with hematological malignancies have TP53 alterations. The highest frequency was observed in acute lymphoblastic leukemia (ALL) (total: 19%; mut+del: 6%; mut only: 8%; del only: 5%) and acute myeloid leukemia (AML) (total: 13%; mut+del: 5%; mut only: 7%; del only: 1%), whereas TP53 alterations occurred less frequently in chronic lymphocytic leukemia (CLL) (total: 8%) and myelodysplastic syndromes (MDS) (total: 7%) [103].
TP53-mutant AML has an extremely poor prognosis and often exhibits natural resistance to chemotherapy [104]. It was found that autophagic flux was higher in poor risk AML compared with favorable- and intermediate-risk AML, although the high autophagy flux related to TP53 mutations, knockdown, or ectopic-overexpressing mutant p53 had no effect on autophagy flux [105]. In addition, in contrast to wtp53 AML, the autophagy inhibitor HCQ treatment did not trigger a BAX and PUMA-dependent apoptotic response in p53mut AMLs. These findings imply that the level of autophagy flux in AML might be an intrinsic property and co-treatment with autophagy inhibitors might only be effective for wtp53 AML patients [105].
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common form of pediatric cancers. As seems to be the case with other types of leukemia, the poor prognostic group of BCR/ABL1-positive BCP-ALL appears particularly dependent on autophagy for their survival and malignant transformation. Exposure of BCP-ALL cells to irradiation triggers autophagy and cell death in a p53-dependent manner [106]. However, the combination with autophagy inhibitors, for which this situation warrants, seems to be related to the mechanism of drug action. Cheong et al. reported that autophagy inhibitors significantly increased sensitivity of the cytarabine arabinoside-resistant U937 cells, which lack the function of p53 [107][108]. Similarly, sorafenib rarely induces autophagy in wtp53 AML cells (OCI-AML3) and p53 null AML cells (HL-60), but induces protective autophagy in p53 null cells (HL-60) [109].
The situations in chronic leukemia are more complicated. Carew and colleges demonstrated that autophagy inhibitors augment the anti-CMLs’ activity of the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) to overcome Bcr-Abl-mediated drug resistance, regardless of p53 status [110]. In the case of another drug, the Src-family protein-tyrosine kinase inhibitor, dasatinib, wtp53 CLL cells are resistant because dasatinib induces cytoprotective autophagy. In contrast, p53 mutant CLL lymphocytes are hypersensitive to dasatinib due to the low level of autophagy [111].
2.2. Gastric Cancer
Helicobacter pylori (HP) is responsible for about 90% of gastric cancer (GC) cases worldwide [112]. Recent work demonstrated that HP promotes p53 proteasomal degradation and inhibits USF1 expression. The low level of USF1 further drives p53 degradation and then accelerates the progression of gastric carcinogenesis, which is related to the low overall survival in GC patients [113]. This reflects the importance of activating p53 in the treatment of gastric cancer. Some p53-regulated proteins also play important roles in regulating autophagy, such as Kallikrein-related peptidase 6 (KLK6), which is p53-dependent and autophagy-related in the tumor microenvironment. Studies have shown that Auranofin, an inhibitor of thioredoxin reductase, induced resistance in gastric cancer cells may be due to overexpression of KLK6, which is affected by p53 upregulation, resulting in protective autophagy [114]. The cdk4/6 inhibitor, Palbociclib, induced p53-dependent autophagy in gastric cancer cells, and the knockdown of p53 was accompanied by a deficiency of the lysosome-mediated degradation of autophagosome cargo, resulting in autophagic blockade [102]. However, the status of p53 does not seem to play a decisive role in some anti-gastric cancer drugs. Tenovin-6 is a potent activator of p53; interestingly, the sensitivity of Tenovin-6 to gastric cancer cell lines and the initiation of autophagy were not correlated with TP53 gene status. Meanwhile, CQ increased Tenovin-6-induced cell death also in a p53-independent manner [115].
2.3. Pancreatic Cancer
Pancreatic cancer is one of the most lethal types of cancer. Most patients with pancreatic cancer have genetic alterations [116], including KRAS [117], TP53 [118], CDKN2A [119], SMAD4 [120], BRCA1, and BRCA2 [121]. Disruptions of KRAS and TP53 are almost universal, with frequencies of about 70–95% [122][123] and 20–76% [124][125], respectively. Autophagy is commonly reported as a protective response for pancreatic cancer cell proliferation in vitro [126][127]. Therefore, CQ or HCQ as a chemical sensitizer (for example, against trametinib, gemcitabine, or nab-paclitaxel) is currently being actively tested in clinical trials (clinical trial: NCT01128296, NCT01506973, NCT01978184). However, these sensitizing effects could not be well reproduced in clinical trials. First, a phase II clinical trial report 5 years ago indicated that in patients with previously treated metastatic pancreatic cancer, HCQ monotherapy demonstrated negligible therapeutic efficacy [128]. Second, a recent phase II clinical trial showed that HCQ did not improve the primary end point of 12-month overall survival in patients with pancreatic cancer treated with first-line drugs, namely gemcitabine hydrochloride and nab-paclitaxel (GA) [129] (NCT01506973). In 2013, Rosenfeldt et al. showed that the status of p53 determines the development of humanized genetically modified mouse models of pancreatic ductal adenocarcinoma. They proved that treatment of mice with the autophagy inhibitor HCQ significantly accelerates tumor formation in mice containing oncogenic Kras but lacking p53 [130]. We questioned whether these unsatisfactory clinical trials could be related to p53 mutations; however, this does not seem to be the case. Yang et al. demonstrated that CQ inhibited the proliferation of pancreatic cancer transplanted tumors, independently of p53 status [131]. Analysis of patients who had undergone combined treatment of HCQ and GA showed that there was no significant correlation between the prognosis of patients after treatment and TP53 mutational status [129]. This suggests that both autophagy inhibitors alone and in combination often produce disappointing results in the clinic, which may not be related to the status of p53. Nevertheless, there have been a few promising outcomes such as a recent study showing that ERK inhibition may enhance the dependence of pancreatic ductal cancer on autophagy [132]. Therefore, blocking the ERK pathway and combining autophagy inhibitors into clinical practice could prove to be an effective strategy in the treatment of pancreatic ductal carcinoma.
2.4. Colorectal Cancer
Colorectal cancer is the third most common cancer in the world and a leading cause of cancer-related deaths [133]. Comprehensive genomic analysis has revealed that p53 mutations exist in about 60% of colorectal cancers, with most being of the missense-type at “hot spots”, which indicates that the mutated p53 has carcinogenic effect through the GOF mechanism [134]. However, the effect of p53 status on chemosensitivity is not consistent. Early studies by Violette et al. found that the resistance of 5-fluorouracil to 8 different kinds of colon cancer cells was related to the relative levels of BCL-2, BCL-x(L), and BAX, but not to the status of p53 [135]. However, clinical studies have shown that colorectal tumors with mutant p53 have a weak or absent response to 5-fluorouracil therapy. Patients with wtp53 colorectal tumors have a longer survival period than those with mutant p53 tumors [136]. This suggests that when chemotherapeutic drugs act on the whole body, p53 may be associated with many other factors, such as tumor microenvironment or autophagy, and the “gatekeeper” role of p53 may be more obvious.
The relationship between p53 status and autophagy in colon cancer has also been studied by the Kroemer laboratory. These investigators reported that knock out p53 in HCT-116 cells improved mouse survival by inducing rather than blocking autophagy [17], and that re-transfection with wtp53 inhibited baseline autophagy [137]. Since these reports, more studies have explored the relationship between the activity of anti-colon cancer agents and the regulation of p53 and autophagy. Compared with HCT-116 p53+/+ cells, HCT-116 p53−/− cells are more sensitive to Crocin (the bioactive molecule of saffron), which is associated with its induction of defective autophagosome formation in HCT-116 p53−/− cells [138]. Betulinic acid (BA), a naturally occurring pentacyclic triterpene, has demonstrated antitumor properties in several human cancers [139]. BA interferes with the induction of protective autophagy by degrading mutant p53 through a ubiquitin-mediated degradation pathway, thus inducing apoptosis and promoting the death of colon cancer cells [140]. The protective autophagy in colon cancer correlated with p53 status may be associated with the loss of the ribosomal protein uL3. Ribosomal protein uL3 has been shown to be a key sensor of nucleolar stress induced by a variety of chemotherapeutic drugs (such as 5-fluorouracil, oxaliplatin, and actinomycin D (Act D)) in p53-deficient colon cancer cells [141][142][143]. Pecoraro et al. further demonstrated that loss of uL3 activated cytoprotective autophagy and in turn mediated resistance to Act D in colon cancer [144]. In addition, Zhang et al. recently proposed that p53 can regulate Ten-eleven-translocation 2 (TET2), a protein that regulates DNA damage by maintaining the DNA repair pathway, through the autophagic degradation pathway. Studies have shown that knockout of TET2 in p53 null colon cancer cells can reverse resistance to chemotherapeutic drugs such as doxorubicin and cisplatin. This provides a potential explanation for drug resistance mechanisms in p53 null colon cancer cells, specifically that loss of p53 leads to lowered degradation of TET2 protein in the cytoplasm, but more accumulation in the nucleus during doxorubicin or cisplatin treatment. Nuclear TET2 protects the genome from DNA damage caused by doxorubicin or cisplatin; ultimately, promoting the growth and survival of colon cancer cells and contributing to chemoresistance [145].
2.5. Liver Cancer
Hepatocellular carcinoma (HCC) is the most common form of liver cancer in adults, accounting for ~85–90% of liver cancer patients [146]. Major risk factors include chronic infections with hepatitis B (HBV) or C (HCV) virus, dietary aflatoxin B1 (AFB1) toxins, or alcohol consumption [147][148]. TP53 mutations are exhibited in ~25–30% of HCC patients [149], more than ~50% in AFB1-related HCC patients, and ~45% of HBV-related HCC patients [150]; thus, detection of point mutations in TP53 is considered a biomarker for AFB1 exposure and risk for HCC. Transversion of G:C to T:A at the third position of codon 249ser was detected in the serum DNA of HCC patient biopsies in areas of high AFB1 exposure and HBV endemic areas [151][152]. This TP53 249ser mutant was shown to inhibit wt p53-mediated apoptosis and facilitate tumor cell growth when transfected into p53 null liver cancer cells [153][154].
HBV is a DNA virus that infects hepatocytes, causing liver injury and hepatocyte cell death, and which can promote tumorigenesis. Its DNA codes for four distinct proteins, envelope protein, nucleocapsid (core) protein, viral reverse transcriptase, and the X gene of HBV (HBx) protein [147]. HBx binds to p53 and decreases p53 binding to XPB [155][156], which is important for nucleotide excision repair interaction between HBx and p53. Furthermore, HBx also inactivates p53-dependent activity, such as p53 mediated transcription of cell cycle regulators, repression of TP53 transcription and p53-activated apoptosis [154][157][158][159][160]. HBx binding to PI3KC3 has also been shown to enhance viral replication by inducing autophagy in hepatoma cells transfected with HBV [161]; furthermore, Mizui et al. demonstrated that autophagy inhibition can limit HCV replication [162]. Furthermore, GOF p53 mutations were demonstrated to alter apoptosis induction in hepatocellular carcinoma. Hep3B cells (p53 null) transfected with varying GOF p53 mutations and p53wt plasmids exhibited an anti-apoptotic gain of function. Mutant p53 was able to repress CD95 (fas/APO-1) gene transcription, as well as repress BAX expression, thus attenuating mitochondrial mediated apoptotic pathways and extrinsic apoptotic pathways [163].
Excessive inflammation and tissue damage due to chronic infection by HBV or HCV or alcohol consumption are major contributors to hepatocarcinogenesis. Autophagy induction can contribute to alleviation of some of this toxic stress by clearing damaged protein accumulation, dysfunctional mitochondria and genomic stress [164]. TAK1-mediated activation of autophagy prevented excessive lipid accumulation, while Tak1-depletion resulted in lipid accumulation, hepatosteatosis, and tumorigenesis [165]. In contrast, high basal autophagy can also promote tumorigenesis and chemoresistance [166][167]. Du et al. demonstrated that autophagy inhibition through ATG7 silencing and CQ pretreatment sensitized HepG2 hepatocarcinoma cells to oxaliplatin treatment and enhanced apoptotic cell death [168].
Although the indicated studies suggest that autophagy is cytoprotective in liver cancer, autophagy has also been shown to play dual functional roles in this disease. Studies by Zhang et al. showed that resveratrol inhibited proliferation and migration of HCC cells through the promotion of autophagy. Furthermore, the anti-tumor effects of resveratrol were attributed to autophagy induction through resveratrol-mediated p53 activation, as well as inhibition of PI3K/AKT [169]. Treatment with 3-MA, an autophagy inhibitor, negated resveratol cytotoxic effects on HCC cell proliferation, invasion and migration. Similarly, Wang et al. demonstrated in HCC cell lines that exposure to fangchinoline, a bisbenzylisoquinoline alkaloid shown to induce cell cycle arrest in breast and prostate cancer cell lines [170], induced autophagy in a p53/sestrin2/AMPK-dependent manner and induced autophagic cell death in HepG2 and PLC/PRF/5 cell lines [171]. Consequently, it is feasible that targeting autophagy may potentiate chemosensitization and induce cell death in hepatocarcinoma cells [172][173].
References
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. Pathol. 2005, 205, 275–292, doi:10.1002/path.1706.
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Rev. Clin. Oncol. 2010, 7, 653–664, doi:10.1038/nrclinonc.2010.139.
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120, doi:1136/gutjnl-2012-302529.
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429, doi:1016/j.ccr.2012.01.007.
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33, doi:1016/j.drup.2015.11.004.
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An "Old" Friend Becomes the Enemy. Cancers 2020, 12, 822, doi:3390/cancers12040822.
- Bargonetti, J.; Manfredi, J.J. Multiple roles of the tumor suppressor p53. Opin. Oncol. 2002, 14, 86–91, doi:10.1097/00001622-200201000-00015.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310, doi:1038/35042675.
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Rev. Cancer 2018, 18, 89–102, doi:10.1038/nrc.2017.109.
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Rev. Clin. Oncol. 2018, 15, 13–30, doi:10.1038/nrclinonc.2017.151.
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Rev. Cancer 2009, 9, 749–758, doi:10.1038/nrc2723.
- Vikhanskaya, F.; Siddique, M.M.; Kei Lee, M.; Broggini, M.; Sabapathy, K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Cancer Res. 2005, 11, 4348–4356, doi:10.1158/1078-0432.CCR-04-1547.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339, doi:1038/nature12634.
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biophys. Acta 2010, 1806, 220–229, doi:10.1016/j.bbcan.2010.07.003.
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651, doi:1158/0008-5472.CAN-13-2966.
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814, doi:4161/auto.6486.
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Cell Biol. 2008, 10, 676–687, doi:10.1038/ncb1730.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078, doi:1016/j.cell.2017.08.028.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431, doi:1016/j.cell.2009.04.037.
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317, doi:1016/j.ccr.2014.01.021.
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221, doi:1126/science.2649981.
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708, doi:1038/342705a0.
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494, doi:1126/science.2554494.
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764, doi:1016/j.ccr.2012.10.022.
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909, doi:1038/cdd.2013.17.
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462, doi:1126/science.1346476.
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. Mol. Cell Biol. 2019, 11, 293–305, doi:10.1093/jmcb/mjy072.
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Cell. Biol. 2005, 25, 10097–10110, doi:10.1128/MCB.25.22.10097-10110.2005.
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.t.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014, 157, 382–394, doi:1016/j.cell.2014.01.066.
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258, doi:1016/j.cell.2011.12.017.
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341, doi:1016/j.cell.2009.11.026.
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91, doi:1016/j.ccr.2011.06.004.
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826, doi:1158/0008-5472.CAN-08-4010.
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Rev. Cancer 2002, 2, 277–288, doi:10.1038/nrc776.
- Kojima, K.; Duvvuri, S.; Ruvolo, V.; Samaniego, F.; Younes, A.; Andreeff, M. Decreased sensitivity of 17p-deleted chronic lymphocytic leukemia cells to a small molecule BCL-2 antagonist ABT-737. Cancer 2012, 118, 1023–1031, doi:1002/cncr.26360.
- Chan, K.T.; Lung, M.L. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother. Pharm. 2004, 53, 519–526, doi:1007/s00280-004-0767-4.
- Wong, R.P.; Tsang, W.P.; Chau, P.Y.; Co, N.N.; Tsang, T.Y.; Kwok, T.T. p53-R273H gains new function in induction of drug resistance through down-regulation of procaspase-3. Cancer Ther. 2007, 6, 1054–1061, doi:10.1158/1535-7163.MCT-06-0336.
- Lozano, G. Restoring p53 in cancer: The promises and the challenges. Mol. Cell Biol. 2019, 11, 615–619, doi:10.1093/jmcb/mjz063.
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665, doi:1038/nature05541.
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. Clin. Investig. 2011, 121, 893–904, doi:10.1172/JCI44504.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660, doi:1038/nature05529.
- Li, Q.; Zhang, Y.; El-Naggar, A.K.; Xiong, S.; Yang, P.; Jackson, J.G.; Chau, G.; Lozano, G. Therapeutic efficacy of p53 restoration in Mdm2-overexpressing tumors. Cancer Res. MCR 2014, 12, 901–911, doi:10.1158/1541-7786.MCR-14-0089.
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. Pathol. 2010, 221, 3–12, doi:10.1002/path.2697.
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90.
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857, doi:1038/s41418-019-0474-7.
- Briceno, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Focus 2003, 14, e3, doi:10.3171/foc.2003.14.2.4.
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Intern. Med. 2006, 144, 337–343, doi:10.7326/0003-4819-144-5-200603070-00008.
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368, doi:4161/auto.28984.
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. J. Mol. Sci. 2017, 18, 1279, doi:10.3390/ijms18061279.
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Rev. Cancer 2012, 12, 401–410, doi:10.1038/nrc3262.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889, doi:1038/nature04724.
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. Cell Biol. 2005, 169, 425–434, doi:10.1083/jcb.200412022.
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. J. Cancer 2009, 124, 1060–1071, doi:10.1002/ijc.24030.
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Commun. 2014, 5, 3056, doi:10.1038/ncomms4056.
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.L.; Liu, Y.; Li, W.F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Cancer 2019, 18, 17, doi:10.1186/s12943-019-0944-z.
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. J. Cancer 2010, 46, 1900–1909, doi:10.1016/j.ejca.2010.02.021.
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99, doi:1038/cdd.2013.124.
- Michaud, M.; Xie, X.; Bravo-San Pedro, J.M.; Zitvogel, L.; White, E.; Kroemer, G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology 2014, 3, e944047, doi:4161/21624011.2014.944047.
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577, doi:1126/science.1208347.
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14, doi:3390/biom8020014.
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053, doi:1158/0008-5472.CAN-06-4149.
- Balaburski, G.M.; Hontz, R.D.; Murphy, M.E. p53 and ARF: Unexpected players in autophagy. Trends Cell Biol. 2010, 20, 363–369, doi:1016/j.tcb.2010.02.007.
- Marchenko, N.D.; Moll, U.M. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle 2007, 6, 1718–1723, doi:4161/cc.6.14.4503.
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014, doi:1126/science.1092734.
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. Biol. Chem. 2006, 281, 8600–8606, doi:10.1074/jbc.M507611200.
- Ulloa, L.; Messmer, D. High-mobility group box 1 (HMGB1) protein: Friend and foe. Cytokine Growth Factor Rev. 2006, 17, 189–201, doi:1016/j.cytogfr.2006.01.003.
- Muller, S.; Ronfani, L.; Bianchi, M.E. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. Intern. Med. 2004, 255, 332–343, doi:10.1111/j.1365-2796.2003.01296.x.
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Immunol. 2018, 38, 40–48, doi:10.1016/j.smim.2018.02.011.
- Ellerman, J.E.; Brown, C.K.; de Vera, M.; Zeh, H.J.; Billiar, T.; Rubartelli, A.; Lotze, M.T. Masquerader: High mobility group box-1 and cancer. Cancer Res. 2007, 13, 2836–2848, doi:10.1158/1078-0432.CCR-06-1953.
- Guo, Z.S.; Liu, Z.; Bartlett, D.L.; Tang, D.; Lotze, M.T. Life after death: Targeting high mobility group box 1 in emergent cancer therapies. J. Cancer Res. 2013, 3, 1–20.
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211, doi:4161/auto.6.8.13651.
- Huang, J.; Liu, K.; Yu, Y.; Xie, M.; Kang, R.; Vernon, P.; Cao, L.; Tang, D.; Ni, J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy 2012, 8, 275–277, doi:4161/auto.8.2.18940.
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J.; 3rd; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005, doi:1158/0008-5472.CAN-11-2291.
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134, doi:1016/j.cell.2006.05.034.
- Lorin, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010, 6, 153–154, doi:4161/auto.6.1.10537.
- Eby, K.G.; Rosenbluth, J.M.; Mays, D.J.; Marshall, C.B.; Barton, C.E.; Sinha, S.; Johnson, K.N.; Tang, L.; Pietenpol, J.A. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Cancer 2010, 9, 95, doi:10.1186/1476-4598-9-95.
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Rev. Cancer 2002, 2, 48–58, doi:10.1038/nrc706.
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Rev. Drug Discov. 2006, 5, 219–234, doi:10.1038/nrd1984.
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76, doi:1007/978-1-60761-416-6_4.
- Ahn, J.H.; Jang, G.H.; Lee, M. Defective autophagy in multidrug resistant cells may lead to growth inhibition by BH3-mimetic gossypol. Cell. Physiol. 2013, 228, 1496–1505, doi:10.1002/jcp.24305.
- Sun, W.L.; Lan, D.; Gan, T.Q.; Cai, Z.W. Autophagy facilitates multidrug resistance development through inhibition of apoptosis in breast cancer cells. Neoplasma 2015, 62, 199–208, doi:4149/neo_2015_025.
- Eom, S.Y.; Hwang, S.H.; Yeom, H.; Lee, M. An ATG5 knockout promotes paclitaxel resistance in v-Ha-ras-transformed NIH 3T3 cells. Biophys. Res. Commun. 2019, 513, 234–241, doi:10.1016/j.bbrc.2019.03.197.
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. J. Cancer 2017, 36, 52, doi:10.1186/s40880-017-0219-2.
- Sirichanchuen, B.; Pengsuparp, T.; Chanvorachote, P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Cell. Biochem. 2012, 364, 11–18, doi:10.1007/s11010-011-1199-1.
- Meschini, S.; Condello, M.; Marra, M.; Formisano, G.; Federici, E.; Arancia, G. Autophagy-mediated chemosensitizing effect of the plant alkaloid voacamine on multidrug resistant cells. Vitr. 2007, 21, 197–203, doi:10.1016/j.tiv.2006.09.007.
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18, doi:3390/cancers10010018.
- Eum, K.H.; Lee, M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH 3T3 cells. Cells 2011, 31, 231–238, doi:10.1007/s10059-011-0034-6.
- Ma, Q.; Chang, Z.; Wang, W.; Wang, B. Rapamycin-Mediated mTOR Inhibition Reverses Drug Resistance to Adriamycin in Colon Cancer Cells. Hepatogastroenterology 2015, 62, 880–886.
- Leisching, G.R.; Loos, B.; Botha, M.H.; Engelbrecht, A.M. The role of mTOR during cisplatin treatment in an in vitro and ex vivo model of cervical cancer. Toxicology 2015, 335, 72–78, doi:1016/j.tox.2015.07.010.
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Oncol. 2016, 10, 1008–1029, doi:10.1016/j.molonc.2016.04.001.
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720, doi:1038/cddis.2013.217.
- Law, B.Y.; Mok, S.W.; Chan, W.K.; Xu, S.W.; Wu, A.G.; Yao, X.J.; Wang, J.R.; Liu, L.; Wong, V.K. Hernandezine, a novel AMPK activator induces autophagic cell death in drug-resistant cancers. Oncotarget 2016, 7, 8090–8104, doi:18632/oncotarget.6980.
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Cell Biol. 2019, 21, 133–142, doi:10.1038/s41556-018-0244-7.
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41, doi:1038/cr.2013.168.
- MacIntyre, A.C.; Cutler, D.J. The potential role of lysosomes in tissue distribution of weak bases. Drug Dispos. 1988, 9, 513–526, doi:10.1002/bod.2510090602.
- Herlevsen, M.; Oxford, G.; Owens, C.R.; Conaway, M.; Theodorescu, D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Cancer Ther. 2007, 6, 1804–1813, doi:10.1158/1535-7163.MCT-06-0372.
- Hrabeta, J.; Belhajova, M.; Subrtova, H.; Merlos Rodrigo, M.A.; Heger, Z.; Eckschlager, T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. J. Mol. Sci. 2020, 21, 4392, doi:10.3390/ijms21124392.
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. Biol. Chem. 2013, 288, 31761–31771, doi:10.1074/jbc.M113.514091.
- Rajagopal, A.; Simon, S.M. Subcellular localization and activity of multidrug resistance proteins. Biol. Cell 2003, 14, 3389–3399, doi:10.1091/mbc.e02-11-0704.
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316.
- Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 80, doi:3390/cells8091080.
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-induced autophagy and senescence in gastric cancer cells. Cell Res. 2017, 360, 390–396, doi:10.1016/j.yexcr.2017.09.031.
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.; Haferlach, C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: An analysis of 3307 cases. Leukemia 2017, 31, 705–711, doi:1038/leu.2016.263.
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121, doi:1182/blood-2011-08-375758.
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927, doi:1038/cddis.2017.317.
- Skah, S.; Richartz, N.; Duthil, E.; Gilljam, K.M.; Bindesboll, C.; Naderi, E.H.; Eriksen, A.B.; Ruud, E.; Dirdal, M.M.; Simonsen, A.; et al. cAMP-mediated autophagy inhibits DNA damage-induced death of leukemia cells independent of p53. Oncotarget 2018, 9, 30434–30449, doi:18632/oncotarget.25758.
- Cheong, J.W.; Kim, Y.; Eom, J.I.; Jeung, H.K.; Min, Y.H. Enhanced autophagy in cytarabine arabinoside-resistant U937 leukemia cells and its potential as a target for overcoming resistance. Med. Rep. 2016, 13, 3433–3440, doi:10.3892/mmr.2016.4949.
- Kanno, S.; Higurashi, A.; Watanabe, Y.; Shouji, A.; Asou, K.; Ishikawa, M. Susceptibility to cytosine arabinoside (Ara-C)-induced cytotoxicity in human leukemia cell lines. Lett. 2004, 152, 149–158, doi:10.1016/j.toxlet.2004.04.014.
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730, doi:3324/haematol.2012.062083.
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322, doi:1182/blood-2006-10-050260.
- Amrein, L.; Soulieres, D.; Johnston, J.B.; Aloyz, R. p53 and autophagy contribute to dasatinib resistance in primary CLL lymphocytes. Res. 2011, 35, 99–102, doi:10.1016/j.leukres.2010.05.029.
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Rev. Cancer 2002, 2, 28–37, doi:10.1038/nrc703.
- Costa, L.; Corre, S.; Michel, V.; Le Luel, K.; Fernandes, J.; Ziveri, J.; Jouvion, G.; Danckaert, A.; Mouchet, N.; Da Silva Barreira, D.; et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 2020, 69, 1582–1591, doi:1136/gutjnl-2019-318640.
- Kim, T.W.; Lee, S.J.; Kim, J.T.; Kim, S.J.; Min, J.K.; Bae, K.H.; Jung, H.; Kim, B.Y.; Lim, J.S.; Yang, Y.; et al. Kallikrein-related peptidase 6 induces chemotherapeutic resistance by attenuating auranofin-induced cell death through activation of autophagy in gastric cancer. Oncotarget 2016, 7, 85332–85348, doi:18632/oncotarget.13352.
- Ke, X.; Qin, Q.; Deng, T.; Liao, Y.; Gao, S.J. Heterogeneous Responses of Gastric Cancer Cell Lines to Tenovin-6 and Synergistic Effect with Chloroquine. Cancers 2020, 12, 365, doi:3390/cancers12020365.
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42, doi:3390/cancers9050042.
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782, doi:1093/nar/16.16.7773.
- Redston, M.S.; Caldas, C.; Seymour, A.B.; Hruban, R.H.; da Costa, L.; Yeo, C.J.; Kern, S.E. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994, 54, 3025–3033.
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130.
- Maurice, D.; Pierreux, C.E.; Howell, M.; Wilentz, R.E.; Owen, M.J.; Hill, C.S. Loss of Smad4 function in pancreatic tumors: C-terminal truncation leads to decreased stability. Biol. Chem. 2001, 276, 43175–43181, doi:10.1074/jbc.M105895200.
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. Clin. Oncol. 2015, 33, 3124–3129, doi:10.1200/JCO.2014.59.7401.
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. J. Cancer 2004, 91, 355–358, doi:10.1038/sj.bjc.6601894.
- Kim, S.T.; Lim, D.H.; Jang, K.T.; Lim, T.; Lee, J.; Choi, Y.L.; Jang, H.L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Cancer Ther. 2011, 10, 1993–1999, doi:10.1158/1535-7163.MCT-11-0269.
- Hwang, R.F.; Gordon, E.M.; Anderson, W.F.; Parekh, D. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene. Surgery 1998, 124, 143–150; discussion 150-141.
- Kern, S.E.; Pietenpol, J.A.; Thiagalingam, S.; Seymour, A.; Kinzler, K.W.; Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992, 256, 827–830, doi:1126/science.1589764.
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819, doi:1111/j.1349-7006.2008.00893.x.
- Hashimoto, D.; Blauer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. J. Cancer 2014, 50, 1382–1390, doi:10.1016/j.ejca.2014.01.011.
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638, doi:1634/theoncologist.2014-0086.
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998, doi:1001/jamaoncol.2019.0684.
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300, doi:1038/nature12865.
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913, doi:1158/2159-8290.CD-14-0362.
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Med. 2019, 25, 628–640, doi:10.1038/s41591-019-0368-8.
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193, doi:3322/caac.21395.
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. Mol. Cell Biol. 2019, 11, 267–276, doi:10.1093/jmcb/mjy075.
- Violette, S.; Poulain, L.; Dussaulx, E.; Pepin, D.; Faussat, A.M.; Chambaz, J.; Lacorte, J.M.; Staedel, C.; Lesuffleur, T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. J. Cancer 2002, 98, 498–504, doi:10.1002/ijc.10146.
- Benhattar, J.; Cerottini, J.P.; Saraga, E.; Metthez, G.; Givel, J.C. p53 mutations as a possible predictor of response to chemotherapy in metastatic colorectal carcinomas. J. Cancer 1996, 69, 190–192, doi:10.1002/(SICI)1097-0215(19960621)69:3<190::AID-IJC7>3.0.CO;2-V.
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061, doi:4161/cc.7.19.6751.
- Amin, A.; Bajbouj, K.; Koch, A.; Gandesiri, M.; Schneider-Stock, R. Defective autophagosome formation in p53-null colorectal cancer reinforces crocin-induced apoptosis. J. Mol. Sci. 2015, 16, 1544–1561, doi:10.3390/ijms16011544.
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid, a natural compound with potent anticancer effects. Anti-Cancer Drugs 2010, 21, 215–227, doi:1097/CAD.0b013e3283357c62.
- Wang, S.; Wang, K.; Zhang, C.; Zhang, W.; Xu, Q.; Wang, Y.; Zhang, Y.; Li, Y.; Zhang, Y.; Zhu, H.; et al. Overaccumulation of p53-mediated autophagy protects against betulinic acid-induced apoptotic cell death in colorectal cancer cells. Cell Death Dis. 2017, 8, e3087, doi:1038/cddis.2017.485.
- Esposito, D.; Crescenzi, E.; Sagar, V.; Loreni, F.; Russo, A.; Russo, G. Human rpL3 plays a crucial role in cell response to nucleolar stress induced by 5-FU and L-OHP. Oncotarget 2014, 5, 11737–11751, doi:18632/oncotarget.2591.
- Pagliara, V.; Saide, A.; Mitidieri, E.; d’Emmanuele di Villa Bianca, R.; Sorrentino, R.; Russo, G.; Russo, A. 5-FU targets rpL3 to induce mitochondrial apoptosis via cystathionine-beta-synthase in colon cancer cells lacking p53. Oncotarget 2016, 7, 50333–50348, doi:18632/oncotarget.10385.
- Russo, A.; Pagliara, V.; Albano, F.; Esposito, D.; Sagar, V.; Loreni, F.; Irace, C.; Santamaria, R.; Russo, G. Regulatory role of rpL3 in cell response to nucleolar stress induced by Act D in tumor cells lacking functional p53. Cell Cycle 2016, 15, 41–51, doi:1080/15384101.2015.1120926.
- Pecoraro, A.; Carotenuto, P.; Franco, B.; De Cegli, R.; Russo, G.; Russo, A. Role of uL3 in the Crosstalk between Nucleolar Stress and Autophagy in Colon Cancer Cells. J. Mol. Sci. 2020, 21, 2143, doi:10.3390/ijms21062143.
- Zhang, J.; Tan, P.; Guo, L.; Gong, J.; Ma, J.; Li, J.; Lee, M.; Fang, S.; Jing, J.; Johnson, G.; et al. p53-dependent autophagic degradation of TET2 modulates cancer therapeutic resistance. Oncogene 2019, 38, 1905–1919, doi:1038/s41388-018-0524-5.
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458, doi:10.1038/nrgastro.2010.100.
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176, doi:1038/sj.onc.1210279.
- Link, T.; Iwakuma, T. Roles of p53 in extrinsic factor-induced liver carcinogenesis. Hepatoma Res. 2017, 3, 95–104, doi:20517/2394-5079.2017.07.
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Genet. 2015, 47, 505–511, doi:10.1038/ng.3252.
- Meng, X.; Franklin, D.A.; Dong, J.; Zhang, Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014, 74, 7161–7167, doi:1158/0008-5472.CAN-14-1446.
- Szymanska, K.; Lesi, O.A.; Kirk, G.D.; Sam, O.; Taniere, P.; Scoazec, J.Y.; Mendy, M.; Friesen, M.D.; Whittle, H.; Montesano, R.; et al. Ser-249TP53 mutation in tumour and plasma DNA of hepatocellular carcinoma patients from a high incidence area in the Gambia, West Africa. J. Cancer 2004, 110, 374–379, doi:10.1002/ijc.20103.
- Kirk, G.D.; Lesi, O.A.; Mendy, M.; Szymanska, K.; Whittle, H.; Goedert, J.J.; Hainaut, P.; Montesano, R. 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene 2005, 24, 5858–5867, doi:1038/sj.onc.1208732.
- Puisieux, A.; Ji, J.; Guillot, C.; Legros, Y.; Soussi, T.; Isselbacher, K.; Ozturk, M. p53-mediated cellular response to DNA damage in cells with replicative hepatitis B virus. Natl. Acad. Sci. USA 1995, 92, 1342–1346, doi:10.1073/pnas.92.5.1342.
- Wang, X.W.; Gibson, M.K.; Vermeulen, W.; Yeh, H.; Forrester, K.; Sturzbecher, H.W.; Hoeijmakers, J.H.; Harris, C.C. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995, 55, 6012–6016.
- Elmore, L.W.; Hancock, A.R.; Chang, S.F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Natl. Acad. Sci. USA 1997, 94, 14707–14712, doi:10.1073/pnas.94.26.14707.
- Ali, A.; Abdel-Hafiz, H.; Suhail, M.; Al-Mars, A.; Zakaria, M.K.; Fatima, K.; Ahmad, S.; Azhar, E.; Chaudhary, A.; Qadri, I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J. Gastroenterol. WJG 2014, 20, 10238–10248, doi:3748/wjg.v20.i30.10238.
- Schaeffer, L.; Roy, R.; Humbert, S.; Moncollin, V.; Vermeulen, W.; Hoeijmakers, J.H.; Chambon, P.; Egly, J.M. DNA repair helicase: A component of BTF2 (TFIIH) basic transcription factor. Science 1993, 260, 58–63, doi:1126/science.8465201.
- Kwon, J.A.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral core protein (HBc) in human liver cells. Chem. 2003, 384, 203–212, doi:10.1515/BC.2003.022.
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Natl. Acad. Sci. USA 1994, 91, 2230–2234, doi:10.1073/pnas.91.6.2230.
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62, doi:1016/s0165-4608(96)00329-9.
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Natl. Acad. Sci. USA 2010, 107, 4383–4388, doi:10.1073/pnas.0911373107.
- Mizui, T.; Yamashina, S.; Tanida, I.; Takei, Y.; Ueno, T.; Sakamoto, N.; Ikejima, K.; Kitamura, T.; Enomoto, N.; Sakai, T.; et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. Gastroenterol. 2010, 45, 195–203, doi:10.1007/s00535-009-0132-9.
- Schilling, T.; Kairat, A.; Melino, G.; Krammer, P.H.; Stremmel, W.; Oren, M.; Muller, M. Interference with the p53 family network contributes to the gain of oncogenic function of mutant p53 in hepatocellular carcinoma. Biophys. Res. Commun. 2010, 394, 817–823, doi:10.1016/j.bbrc.2010.03.082.
- Yang, S.; Yang, L.; Li, X.; Li, B.; Li, Y.; Zhang, X.; Ma, Y.; Peng, X.; Jin, H.; Li, H. New insights into autophagy in hepatocellular carcinoma: Mechanisms and therapeutic strategies. J. Cancer Res. 2019, 9, 1329–1353.
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. Clin. Investig. 2014, 124, 3566–3578, doi:10.1172/JCI74068.
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470, doi:1101/gad.2016311.
- Song, J.; Qu, Z.; Guo, X.; Zhao, Q.; Zhao, X.; Gao, L.; Sun, K.; Shen, F.; Wu, M.; Wei, L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009, 5, 1131–1144, doi:4161/auto.5.8.9996.
- Du, H.; Yang, W.; Chen, L.; Shi, M.; Seewoo, V.; Wang, J.; Lin, A.; Liu, Z.; Qiu, W. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Rep. 2012, 27, 143–150, doi:10.3892/or.2011.1464.
- Zhang, B.; Yin, X.; Sui, S. Resveratrol inhibited the progression of human hepatocellular carcinoma by inducing autophagy via regulating p53 and the phosphoinositide 3kinase/protein kinase B pathway. Rep. 2018, 40, 2758–2765, doi:10.3892/or.2018.6648.
- Liu, Y.; Xia, B.; Lan, J.; Hu, S.; Huang, L.; Chen, C.; Zeng, X.; Lou, H.; Lin, C.; Pan, W. Design, Synthesis and Anticancer Evaluation of Fangchinoline Derivatives. Molecules 2017, 22, 1923, doi:3390/molecules22111923.
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. J. Pharmacol. 2011, 164, 731–742, doi:10.1111/j.1476-5381.2011.01349.x.
- Schaper, L.A.; Ofele, K.; Kadyrov, R.; Bechlars, B.; Drees, M.; Cokoja, M.; Herrmann, W.A.; Kuhn, F.E. N-Heterocyclic carbenes via abstraction of ammonia: ‘normal’ carbenes with ‘abnormal’ character. Commun. 2012, 48, 3857–3859, doi:10.1039/c2cc30611e.
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172, doi:4161/auto.7.10.16818.

