Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Badran | -- | 2222 | 2022-05-31 18:11:02 | | | |
| 2 | Rita Xu | -7 word(s) | 2215 | 2022-06-01 04:03:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Badran, M.; Gozal, D. Plasminogen Activator Inhibitor-1. Encyclopedia. Available online: https://encyclopedia.pub/entry/23617 (accessed on 23 July 2026).
Badran M, Gozal D. Plasminogen Activator Inhibitor-1. Encyclopedia. Available at: https://encyclopedia.pub/entry/23617. Accessed July 23, 2026.
Badran, Mohammad, David Gozal. "Plasminogen Activator Inhibitor-1" Encyclopedia, https://encyclopedia.pub/entry/23617 (accessed July 23, 2026).
Badran, M., & Gozal, D. (2022, May 31). Plasminogen Activator Inhibitor-1. In Encyclopedia. https://encyclopedia.pub/entry/23617
Badran, Mohammad and David Gozal. "Plasminogen Activator Inhibitor-1." Encyclopedia. Web. 31 May, 2022.
Copy Citation
Plasminogen activator inhibitor-1 (PAI-1), the primary inhibitor of tissue-type plasminogen activator (tPA) and urinary-type plasminogen activator (uPA), is a key regulator of fibrinolysis and cell migration. Indeed, elevated PAI-1 expression is associated with major cardiovascular adverse events that have been attributed to its antifibrinolytic activity. However, extensive evidence indicates that PAI-1 can induce endothelial dysfunction and atherosclerosis through complex interactions within the vasculature in an antifibrinolytic-independent matter.
obstructive sleep apnea
intermittent hypoxia
plasminogen activator inhibitor-1
atherosclerosis
endothelial dysfunction
1. Introduction
Obstructive sleep apnea (OSA) is a chronic condition affecting up to one billion people worldwide [1]. OSA is defined as a sleep-breathing disorder that involves a decrease or complete cessation of airflow despite ongoing efforts to breathe due to a collapsed upper airway. This leads to partial reductions (hypopneas) and complete pauses (apneas) in breathing that usually last between 10 and 30 s, but some may persist longer. This can lead to abrupt reductions in blood oxygen saturation, with oxygen levels falling as much as 40% or more in severe cases [2]. As a result, several pathological mechanisms ensue such as intermittent hypoxia (IH), sleep fragmentation, episodic hypercapnia, and increased intrathoracic pressure swings [3][4][5]. Consequently, these processes can induce major changes in the autonomic nervous system balance with both increased tonic and reactive sympathetic activity along with parasympathetic withdrawal, disruption of the hypothalamic–pituitary–adrenal-axis, systemic and cellular oxidative stress, and inflammation, fibrosis, and accelerated cellular senescence, all of which resulted in neurocognitive deficits, endothelial dysfunction, hypertension, and atherosclerosis [6][7][8][9][10][11][12][13]. Predictably, OSA is considered as an independent risk factor for cardiovascular disease (CVD) including coronary artery disease (CAD), ischemic stroke, and myocardial infarction (MI) [14]. The majority of strokes and MIs seem to be prompted by atherothrombotic events along with compromised fibrinolytic activity, increasing the propensity for such events [15][16]. The fibrinolytic system is designed to cleave the insoluble polymeric network of fibrin from the vascular system to prevent clot overgrowth and vessel occlusion. Generally, plasminogen is activated by serine proteases plasminogen activators (PAs) including tissue-type PA (tPA) and urokinase-type PA (uPA) into plasmin, which in turn lyses the fibrin and other extracellular matrix components [17][18]. To prevent bleeding, plasminogen activator inhibitor-1 (PAI-1) is normally synthesized in equimolar amounts to PAs, forms a covalent bond with Pas, and stabilizes fibrin [19]. However, many processes including oxidative stress [20], inflammation [21], and fibrosis [22] can lead to elevated levels of PAI-1, which have been implicated in a multitude of diseases and conditions including CVD [23], cancer [24], metabolic disease [25], renal disease [26], behavioral and psychiatric conditions [27], and aging processes [28]. Furthermore, PAI-1 has been shown to induce endothelial dysfunction and atherosclerosis through antifibrinolytic-dependent mechanisms including inflammation [29], endothelial nitric oxide synthase (eNOS) inhibition [30], neointimal hyperplasia [31], and vascular senescence [28]. Despite the fact that PAI-1 levels are consistently elevated in OSA patients [32][33][34][35][36][37][38][39][40][41][42], and that OSA can trigger processes that can upregulate PAI-1 production, little to no attention has been given to PAI-1 as a biomarker or as a promoter of OSA-induced CVD in clinical practice.
2. PAI-1 Sources, Structure, and Function
PAI-1 can be synthetized by numerous types of cells including platelets, macrophages, adipocytes, hepatocytes, vascular smooth muscle cells, endothelial cells, and others [43][44][45]. Approximately 10% of the PAI-1 produced circulates in the blood or is deposited in the subendothelial matrix, while the rest is retained in platelets [46][47]. Platelets can de novo synthesize PAI-1 despite lacking nuclei through activated PAI-1 mRNA, with the synthesis rates being increased upon platelet activation [48]. The circulating PAI-1 fraction exists in its active conformation at levels of 5–50 ng/mL with large intra- and inter-personal variability, while platelet PAI-1 concentrations can reach up to 300 ng/mL with 50% shown to be biologically active [46][49][50]. Ultimately, PAI-1 plasma levels are increased under numerous pathological conditions [51]. The structure and function of PAI-1 have been extensively reviewed previously [25][52][53]. Briefly, PAI-1 is a single chain molecule with two interactive domains including a surface-exposed reactive center loop (RCL) that presents as a substrate peptide becoming the primary site for uPA/tPA binding, and a flexible joint region with helices D, E, and F that bind to vitronectin and stabilize PAI-1 in its active form while enhancing its binding affinity to uPA/tPA 200-fold [24][54][55][56][57][58][59]. PAI-1 exists in three distinct structurally and functionally distinct conformations including active, latent, and cleaved [52][60]. Unless bound to vitronectin, the active form can be readily converted to the more energetically favorable inactive latent form by internalizing the RCL, which may serve as a regulatory mechanism to prevent excessive anti-fibrinolysis [61][62][63]. However, the latent form can be reactivated. In its cleaved form, PAI-1 is still able to bind to other proteins with its helix, but its ability to inhibit uPA/tPA is abrogated [61]. As alluded to earlier, PAI-1 is a master regulator of the plasminogen system. PAI-1 can rapidly inactivate uPA/tPA with a second-order-rate constant between 106 and 107 m−1 s−1, forming a non-covalent Michaelis-like complex and eventually forming an ester bond between the serine residue of the protease and the carboxyl group of the P1 residue of PAI-1 [64][65]. PAI-1 also plays an important role in extracellular matrix (ECM) remodeling by indirectly modulating the activity of matrix metalloproteinases (MMPs) [66]. Indeed, by inhibiting the plasmin activation required for the cleavage of pro-MMP, PAI-1 can block ECM degradation [53].
3. Mechanisms Involved in PAI-1 Upregulation
The human PAI-1 promoter shows a high degree of homology with mice and rats, suggesting that they are regulated by similar mechanisms. The 5′-flanking region contains a ‘TATA’ box with several transcription binding sites including hypoxia inducible factor-1α (HIF-1α), Smads, activator protein-1 (AP-1), specificity protein-1 (SP-1), and nuclear factor kappa B (NF-ƘB). The major contributors to PAI-1 upregulation (Figure 1).
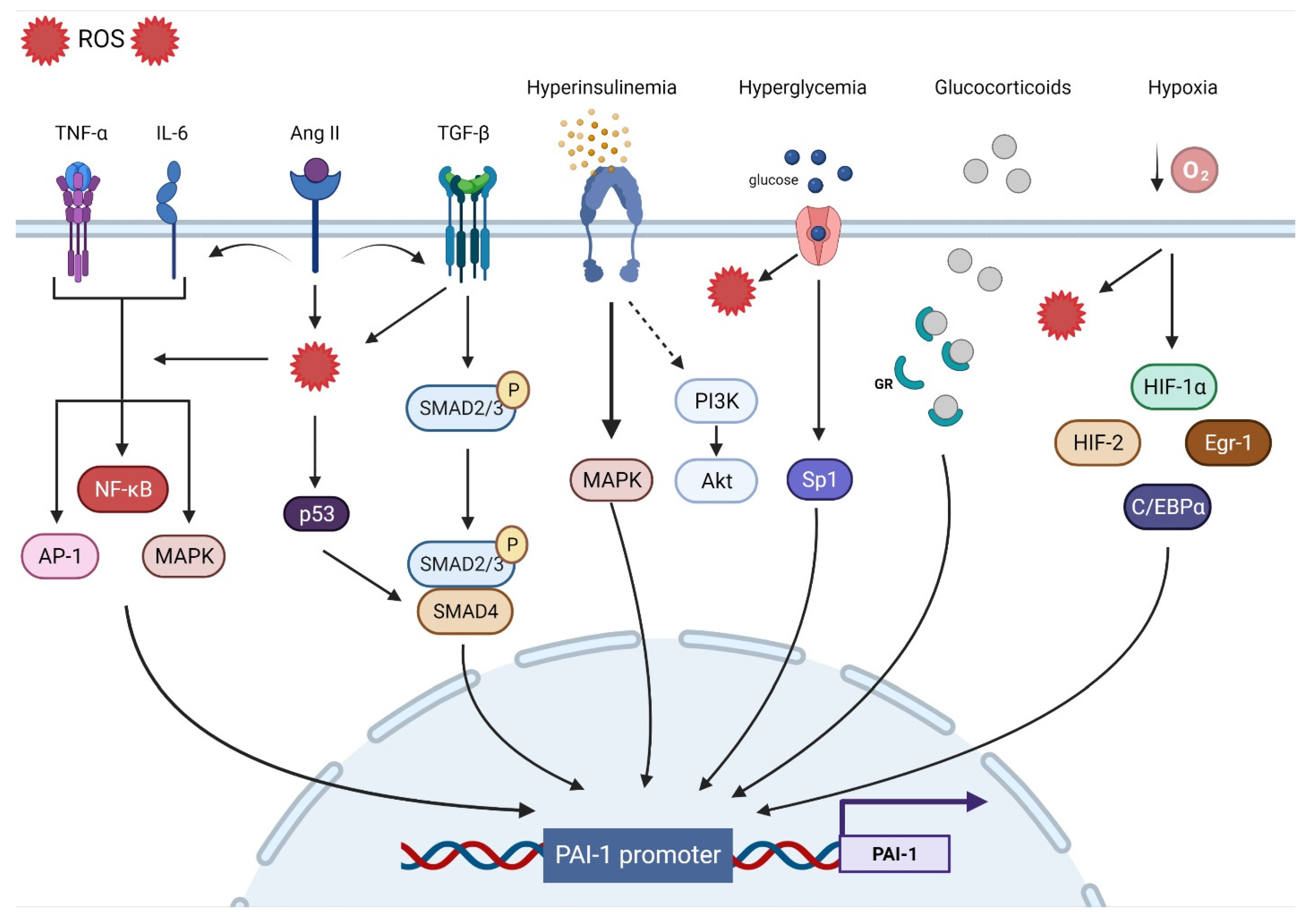
Figure 1. Transcriptional regulatory pathways implicated in PAI-1 synthesis.
3.1. Oxidative Stress
Oxidative stress (OS) is the end result of an imbalance between the production of oxidants and the capacity of the antioxidant system. Although they play an important role in regulating cellular function and signal transduction, free radicals such as reactive oxygen species (ROS) can be detrimental when produced in excess, given their ability to damage lipids, proteins, and DNA [67]. OS is undeniably a major contributor to multiorgan dysfunction in many disease states including CVD [68]. Indeed, ROS overproduction directly decreases nitric oxide (NO) bioavailability, uncouples eNOS, oxidizes low-density lipoprotein (OxLDL), and induces vascular inflammation [69]. OS is a significant upregulator of PAI-1 transcription. Indeed, incubating endothelial cells with H2O2 induced marked increases in PAI-1 mRNA and protein expression [70]. Conversely, the PAI-1 promoter was suppressed by up to 75% in the presence of antioxidants [71]. Furthermore, inhibiting NADPH oxidase, a major source of ROS, abolished the PAI-1 release and promoter activity in cultured endothelial cells [72]. Other experimental in vitro and in vivo studies performed in animal models as well as in humans have shown that the administration of antioxidants can decrease PAI-1 expression [20][73][74][75][76][77][78][79][80][81]. Due to their intricate interactions with multiple signaling pathways and transcription factors, ROS are involved in most of the mechanisms regulating PAI-1 expression. For instance, ROS-induced PAI-1 increased transcription and expression is mediated through the activation of mitogen-activated protein kinase (MAPK) and NF-ƘB pathways that are tightly involved in pro-fibrotic and pro-inflammatory pathways [72][82]. ROS signaling can also stimulate AP-1, HIF-1α, and p53, all of which can increase the transcription of PAI-1 [83][84] (Figure 1).
3.2. Inflammation
Inflammation is a complex constellation of reactions between the host normal defense processes to internal and external stressors that have been implicated in many conditions and age-related diseases, especially in promoting atherosclerosis, a hallmark of CVD [85][86][87]. Low-grade inflammation induces endothelial dysfunction and subintimal cholesterol accumulation, leading to the upregulation of intercellular adhesion molecules and selectins that promote the binding and transmigration of inflammatory cells including monocytes and T-helper cells into the vessel wall. Infiltrating monocytes can transform into resident macrophages that express and activate inflammasomes that are key to the propagation of inflammation through the generation of multiple cytokines that amplify the inflammatory cascade within the vessel wall [87]. Coupled with enhanced ROS production, inflammation enters a vicious cycle in combination with OS, further aggravating atherosclerosis [88]. The link between inflammation and the fibrinolytic system is well-established. Experimental in vitro and in vivo studies as well as clinical studies have identified tumor necrosis factor-α (TNF-α) as a substantial contributor to increased PAI-1 expression [89][90][91][92][93][94]. In endothelial cells, TNF-α upregulated PAI-1 levels and was abolished by N-acetyl cysteine, indicating ROS as a mediator [71]. Administration of TNF-α in mice significantly increased the PAI-1 levels in adipose tissue, while obese mice treated with antibodies targeting TNF-α exhibited reduced plasma PAI-1 expression and adipose tissue-PAI-1 levels [95][96]. It is suggested that TNF-α can induce PAI-1 gene expression via redox-sensitive mechanisms triggering NF-ƘB translocation and interaction with a regulatory region that is present on the PAI-1 promoter [21][94]. These data showcase the interplay between inflammation and OS and their integral role in upregulating PAi-1. Other pathways have been suggested in TNF-α-mediated PAI-1 induction including MAPK and protein kinase C [91]. Interleukin-6 (IL-6) is another inflammatory cytokine involved in PAI-1 upregulation. IL-6 is an acute phase inflammatory reaction protein that can induce C-reactive protein (CRP) synthesis and cortisol production [97]. Animals injected with IL-6 had significant increases in PAI-1 levels, while using IL-6 receptor antagonist reduced the PAI-1 expression in COVID patients [98][99]. IL-6 can activate NF-ƘB and MAPK, leading to increased PAI-1 transcription [53][100] (Figure 1).
3.3. Fibrosis
Progressive vascular fibrosis is a prominent feature of atherosclerosis and CVD [101]. Transforming growth factor-β (TGF-β) is a major regulator of the fibroproliferative response to tissue damage [102]. TGF-β can control cell proliferation and migration, matrix synthesis, calcification, and immunomodulation, all being integral components of atherosclerosis [103]. TGF-β can be produced by all cells composing the vasculature and can also be produced in atherosclerotic lesions. However, TGF-β is mainly released by activated platelets adherent to activated endothelium. As a result, TGF-β induces the transcription of platelet-derived growth factor, collagens, fibronectin, and thrombospondins while suppressing the breakdown of ECM by inducing the transcription of PAI-1 and metalloprotease inhibitors, leading to the accumulation of the fibrotic matrix followed by calcification [101][103]. Overall, TGF-β production in atherosclerotic lesions can result in negative remodeling and progressive narrowing of the arteries, leading to MI and stroke [101]. TGF-β is considered as one of the major drivers of PAI-1 upregulation. In vitro studies have shown that PAI-1 expression is induced by TGF-β in various types of cells, while elevated PAI-1 levels are associated with enhanced TGF-β expression and ECM deposition under many pathological conditions [22][104][105][106][107][108][109]. TGF-β can induce PAI-1 production through the activation of the Smad pathway via the nuclear translocation of the Smad 2/3 and Smad 4 complex and binding to the PAI-1 promoter [110]. Interestingly, TGF-β can induce ROS production and suppress antioxidant activity in various types of cells and in vivo [111][112][113][114][115][116][117]. Thus, PAI-1 expression can also be mediated through TGF-β-induced ROS production. MAPK and NF-ƘB signaling are redox sensitive pathways that can be induced by TGF-β [53][112][118][119]. In TGF-β treated cells, inhibition of NADPH oxidase blocked TGF-β induced MAPK activated PAI-1 expression [83]. Furthermore, TGF-β can upregulate PAI-1 through Smad interactions with p53 and the transcription factors AP-1 and SP-1 [22][83][120] (Figure 1).
3.4. Hypoxia
Hypoxia triggers many cellular processes both in physiological and pathological conditions and has been associated with vascular dysfunction and atherosclerosis [121]. Vascular wall cells respond to hypoxia by tuning metabolism, angiogenesis, inflammation, cell survival signaling, and ultimately, may develop endothelial dysfunction [122][123]. The main regulator of such processes is the transcription factor HIF-1α. Under normoxic conditions, HIF-1α is constantly degraded, whereas hypoxia promotes its stability and transcriptional activity [124]. However, HIF-1α is stabilized in atherosclerotic lesions even under normoxic conditions. ROS, OxLDL, NF-ƘB, and other factors are promoted by HIF-1α and in return, enhance HIF-1α stability [121]. PAI-1 is one of the main transcriptional targets of HIF-1α. Indeed, cells exposed to hypoxia display increased PAI-1 mRNA expression and stability [125][126][127][128][129]. HIF-1α knockdown limited irradiation-induced PAI-1 upregulation in endothelial cells [130]. ROS production in endothelial cells induced HIF-1α and subsequently PAI-1 production [131][132]. Additionally, ROS induced HIF-1α via a specific NF-ƘB binding site in the HIF-1 promoter [133]. Indeed, upregulation of the pulmonary artery smooth muscle PAI-1 was induced by an NF-ƘB-dependent HIF-1α transcription [134]. Although HIF-1α appears to dominate the PAI-1 transcriptional response to hypoxia, other pathways including HIF-2α, early growth response protein-1 (Egr-1), and CCAAT-enhancer-binding protein-α (C/EBPα) can augment this response independently of HIF-1α [135][136] (Figure 1).
3.5. Hormones
Insulin can directly stimulate PAI-1 production in hepatocytes, an effect that is augmented by the presence of insulin-like growth factor [137][138]. The same effect was observed in cocultured endothelial cells and smooth muscle cells (SMCs) [139]. In the context of insulin resistance, compensatory hyperinsulinemia decreases the activity of the PI3-K/Akt pathway and augments the MAPK/ERK pathway, a major driver of PAI-1 production [140][141]. Elevated levels of glucose can also directly increase the expression of PAI-1 in endothelial cells and SMC through an effect on two adjacent Sp1 sites [120]. These data explain the elevated levels of PAI-1 in conditions characterized by hyperinsulinemia and hyperglycemia such as obesity, metabolic syndrome, and type 2 diabetes mellitus [25][142][143]. Under intense stress, very high levels of glucocorticoid hormones can increase the production of PAI-1 protein [144]. Glucocorticoids bind to their cytoplasmic glucocorticoid receptor and the complex is translocated to the nucleus and directly binds to the glucocorticoid response element that enhances PAI-1 transcription [84]. Angiotensin II, a major vasoconstrictor and contributor to hypertension upregulated by the activation of the renin–angiotensin–aldosterone system (RAAS), has been reported to induce PAI-1 expression in cultured endothelial cells in an angiotensin receptor independent manner [145]. Ang II can increase ROS production, fibrotic signaling (TGF-β), and inflammation, all of which can increase the expression of PAI-1 [146][147][148] (Figure 1).
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.L.D.; et al. Estimation of the Global Prevalence and Burden of Obstructive Sleep Apnoea: A Literature-Based Analysis. Lancet Respir. Med. 2019, 7, 687–698.
- Kapur, V.K.; Auckley, D.H.; Chowdhuri, S.; Kuhlmann, D.C.; Mehra, R.; Ramar, K.; Harrod, C.G. Clinical Practice Guideline for Diagnostic Testing for Adult Obstructive Sleep Apnea: An American Academy of Sleep Medicine Clinical Practice Guideline. J. Clin. Sleep Med. 2017, 13, 479–504.
- Badran, M.; Yassin, B.A.; Fox, N.; Laher, I.; Ayas, N. Epidemiology of Sleep Disturbances and Cardiovascular Consequences. Can. J. Cardiol. 2015, 31, 873–879.
- Golbidi, S.; Badran, M.; Ayas, N.; Laher, I. Cardiovascular Consequences of Sleep Apnea. Lung 2012, 190, 113–132.
- Badran, M.; Ayas, N.; Laher, I. Insights into Obstructive Sleep Apnea Research. Sleep Med. 2014, 15, 485–495.
- Badran, M.; Yassin, B.A.; Lin, D.T.S.; Kobor, M.S.; Ayas, N.; Laher, I. Gestational Intermittent Hypoxia Induces Endothelial Dysfunction, Reduces Perivascular Adiponectin and Causes Epigenetic Changes in Adult Male Offspring. J. Physiol. 2019, 597, 5349–5364.
- Badran, M.; Abuyassin, B.; Golbidi, S.; Ayas, N.; Laher, I. Uncoupling of Vascular Nitric Oxide Synthase Caused by Intermittent Hypoxia. Oxid. Med. Cell. Longev. 2016, 2354870.
- Badran, M.; Golbidi, S.; Devlin, A.; Ayas, N.; Laher, I. Chronic Intermittent Hypoxia Causes Endothelial Dysfunction in a Mouse Model of Diet-Induced Obesity. Sleep Med. 2014, 15, 596–602.
- Castro-Grattoni, A.L.; Alvarez-Buvé, R.; Torres, M.; Farré, R.; Montserrat, J.M.; Dalmases, M.; Almendros, I.; Barbé, F.; Sánchez-De-La-Torre, M. Intermittent Hypoxia-Induced Cardiovascular Remodeling Is Reversed by Normoxia in a Mouse Model of Sleep Apnea. Chest 2016, 149, 1400–1408.
- Trzepizur, W.; Cortese, R.; Gozal, D. Murine Models of Sleep Apnea: Functional Implications of Altered Macrophage Polarity and Epigenetic Modifications in Adipose and Vascular Tissues. Metabolism 2018, 84, 44–55.
- Carreras, A.; Kayali, F.; Zhang, J.; Hirotsu, C.; Wang, Y.; Gozal, D. Metabolic Effects of Intermittent Hypoxia in Mice: Steady versus High-Frequency Applied Hypoxia Daily during the Rest Period. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R700–R709.
- Pollicina, I.; Maniaci, A.; Lechien, J.R.; Iannella, G.; Vicini, C.; Cammaroto, G.; Cannavicci, A.; Magliulo, G.; Pace, A.; Cocuzza, S.; et al. Neurocognitive Performance Improvement after Obstructive Sleep Apnea Treatment: State of the Art. Behav. Sci. 2021, 11, 180.
- Seda, G.; Han, T.S. Effect of Obstructive Sleep Apnea on Neurocognitive Performance. Sleep Med. Clin. 2020, 15, 77–85.
- Tietjens, J.R.; Claman, D.; Kezirian, E.J.; de Marco, T.; Mirzayan, A.; Sadroonri, B.; Goldberg, A.N.; Long, C.; Gerstenfeld, E.P.; Yeghiazarians, Y. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J. Am. Heart Assoc. 2019, 8, e010440.
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472.
- Juhan-Vague, I.; Pyke, S.D.M.; Alessi, M.C.; Jespersen, J.; Haverkate, F.; Thompson, S.G. Fibrinolytic Factors and the Risk of Myocardial Infarction or Sudden Death in Patients with Angina Pectoris. Circulation 1996, 94, 2057–2063.
- Rijken, D.C.; Lijnen, H.R. New Insights into the Molecular Mechanisms of the Fibrinolytic System. J. Thromb. Haemost. 2009, 7, 4–13.
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the Control of Blood Coagulation. Blood Rev. 2015, 29, 17.
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen Activator Inhibitor 1: Physiological and Pathophysiological Roles. News Physiol. Sci. 2002, 17, 56–61.
- Eun, A.L.; Ji, Y.S.; Jiang, Z.; Mi, R.Y.; Min, K.K.; Ha, H.; Hi, B.L. Reactive Oxygen Species Mediate High Glucose–Induced Plasminogen Activator Inhibitor-1 up-Regulation in Mesangial Cells and in Diabetic Kidney. Kidney Int. 2005, 67, 1762–1771.
- Swiatkowska, M.; Szemraj, J.; Cierniewski, C.S. Induction of PAI-1 Expression by Tumor Necrosis Factor Alpha in Endothelial Cells Is Mediated by Its Responsive Element Located in the 4G/5G Site. FEBS J. 2005, 272, 5821–5831.
- Guo, B.; Inoki, K.; Isono, M.; Mori, H.; Kanasaki, K.; Sugimoto, T.; Akiba, S.; Sato, T.; Yang, B.; Kikkawa, R.; et al. MAPK/AP-1-Dependent Regulation of PAI-1 Gene Expression by TGF-Beta in Rat Mesangial Cells. Kidney Int. 2005, 68, 972–984.
- Song, C.; Burgess, S.; Eicher, J.D.; O’Donnell, C.J.; Johnson, A.D.; Huang, J.; Sabater-Lleal, M.; Asselbergs, F.W.; Tregouet, D.; Shin, S.Y.; et al. Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J. Am. Heart Assoc. 2017, 6, e004918.
- Placencio, V.R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 in Cancer: Rationale and Insight for Future Therapeutic Testing. Cancer Res. 2015, 75, 2969–2974.
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170.
- MaŁgorzewicz, S.; Skrzypczak-Jankun, E.; Jankun, J. Plasminogen Activator Inhibitor-1 in Kidney Pathology (Review). Int. J. Mol. Med. 2013, 31, 503–510.
- Jiang, H.; Li, X.; Chen, S.; Lu, N.; Yue, Y.; Liang, J.; Zhang, Z.; Yuan, Y. Plasminogen Activator Inhibitor-1 in Depression: Results from Animal and Clinical Studies. Sci. Rep. 2016, 6, 30464.
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. PAI-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446.
- Praetner, M.; Zuchtriegel, G.; Holzer, M.; Uhl, B.; Schaubächer, J.; Mittmann, L.; Fabritius, M.; Fürst, R.; Zahler, S.; Funken, D.; et al. Plasminogen Activator Inhibitor-1 Promotes Neutrophil Infiltration and Tissue Injury on Ischemia-Reperfusion. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 829–842.
- Garcia, V.; Park, E.J.; Siragusa, M.; Frohlich, F.; Haque, M.M.; Pascale, J.V.; Heberlein, K.R.; Isakson, B.E.; Stuehr, D.J.; Sessa, W.C. Unbiased Proteomics Identifies Plasminogen Activator Inhibitor-1 as a Negative Regulator of Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 2020, 117, 9497–9507.
- Ji, Y.; Weng, Z.; Fish, P.; Goyal, N.; Luo, M.; Myears, S.P.; Strawn, T.L.; Chandrasekar, B.; Wu, J.; Fay, W.P. Pharmacological Targeting of Plasminogen Activator Inhibitor-1 Decreases Vascular Smooth Muscle Cell Migration and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2167–2175.
- Von Känel, R.; Natarajan, L.; Ancoli-Israel, S.; Mills, P.J.; Loredo, J.S.; Dimsdale, J.E. Day/Night Rhythm of Hemostatic Factors in Obstructive Sleep Apnea. Sleep 2010, 33, 371–377.
- Gileles-Hillel, A.; Alonso-Álvarez, M.L.; Kheirandish-Gozal, L.; Peris, E.; Cordero-Guevara, J.A.; Terán-Santos, J.; Martinez, M.G.; Jurado-Luque, M.J.; Corral-Peñafiel, J.; Duran-Cantolla, J.; et al. Inflammatory Markers and Obstructive Sleep Apnea in Obese Children: The NANOS Study. Mediators Inflamm. 2014, e605280.
- Zakrzewski, M.; Zakrzewska, E.; Kiciński, P.; Przybylska-Kuć, S.; Dybała, A.; Myśliński, W.; Pastryk, J.; Tomaszewski, T.; Mosiewicz, J. Evaluation of Fibrinolytic Inhibitors: Alpha-2-Antiplasmin and Plasminogen Activator Inhibitor 1 in Patients with Obstructive Sleep Apnoea. PLoS ONE 2016, 11, 166725.
- Nizankowska-Jȩdrzejczyk, A.; Almeida, F.R.; Lowe, A.A.; Kania, A.; Nastałek, P.; Mejza, F.; Foley, J.H.; Nizankowska-Mogilnicka, E.; Undas, A. Modulation of Inflammatory and Hemostatic Markers in Obstructive Sleep Apnea Patients Treated with Mandibular Advancement Splints: A Parallel, Controlled Trial. J. Clin. Sleep Med. 2014, 10, 255–262.
- Bagai, K.; Muldowney, J.A.S.; Song, Y.; Wang, L.; Bagai, J.; Artibee, K.J.; Vaughan, D.E.; Malow, B.A. Circadian Variability of Fibrinolytic Markers and Endothelial Function in Patients with Obstructive Sleep Apnea. Sleep 2014, 37, 359–367.
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Dimsdale, J.E. Association between Sleep Apnea Severity and Blood Coagulability: Treatment Effects of Nasal Continuous Positive Airway Pressure. Sleep Breath. 2006, 10, 139–146.
- Phillips, C.L.; McEwen, B.J.; Morel-Kopp, M.C.; Yee, B.J.; Sullivan, D.R.; Ward, C.M.; Tofler, G.H.; Grunstein, R.R. Effects of Continuous Positive Airway Pressure on Coagulability in Obstructive Sleep Apnoea: A Randomised, Placebo-Controlled Crossover Study. Thorax 2012, 67, 639–644.
- Rangemark, C.; Hedner, J.A.; Carlson, J.T.; Gleerup, G.; Winther, K. Platelet Function and Fibrinolytic Activity in Hypertensive and Normotensive Sleep Apnea Patients. Sleep 1995, 18, 188–194.
- Kheirandish-Gozal, L.; Gileles-Hillel, A.; Alonso-Álvarez, M.L.; Peris, E.; Bhattacharjee, R.; Terán-Santos, J.; Duran-Cantolla, J.; Gozal, D. Effects of Adenotonsillectomy on Plasma Inflammatory Biomarkers in Obese Children with Obstructive Sleep Apnea: A Community-Based Study. Int. J. Obes. 2015, 39, 1094–1100.
- Martin, R.A.; Strosnider, C.; Giersch, G.; Womack, C.J.; Hargens, T.A. The Effect of Acute Aerobic Exercise on Hemostasis in Obstructive Sleep Apnea. Sleep Breath. 2017, 21, 623–629.
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Mills, P.J.; Dimsdale, J.E. Elevated Plasminogen Activator Inhibitor 1 in Sleep Apnea and Its Relation to the Metabolic Syndrome: An Investigation in 2 Different Study Samples. Metabolism 2007, 56, 969–976.
- Simpson, A.J.; Booth, N.A.; Moore, N.R.; Bennett, B. Distribution of Plasminogen Activator Inhibitor (PAI-1) in Tissues. J. Clin. Pathol. 1991, 44, 139–143.
- Crandall, D.L.; Quinet, E.M.; Morgan, G.A.; Busler, D.E.; Mchendry-Rinde, B.; Kral, J.G. Synthesis and Secretion of Plasminogen Activator Inhibitor-1 by Human Preadipocytes. J. Clin. Endocrinol. Metab. 1999, 84, 3222–3227.
- Zhang, L.; Seiffert, D.; Fowler, B.J.; Jenkins, G.R.; Thinnes, T.C.; Loskutoff, D.J.; Parmer, R.J.; Miles, L.A. Plasminogen Has a Broad Extrahepatic Distribution. Thromb. Haemost. 2002, 87, 493–501.
- Booth, N.A.; Simpson, A.J.; Croll, A.; Bennett, B.; MacGregor, I.R. Plasminogen Activator Inhibitor (PAI-1) in Plasma and Platelets. Br. J. Haematol. 1988, 70, 327–333.
- Charlton, P. The Status of Plasminogen Activator Inhibitor-1 as a Therapeutic Target. Expert Opin. Investig. Drugs 1997, 6, 539–554.
- Brogren, H.; Wallmark, K.; Deinum, J.; Karlsson, L.; Jern, S. Platelets Retain High Levels of Active Plasminogen Activator Inhibitor 1. PLoS ONE 2011, 6, e26762.
- Torr-Brown, S.R.; Sobel, B.E. Attenuation of Thrombolysis by Release of Plasminogen Activator Inhibitor Type-1 from Platelets. Thromb. Res. 1993, 72, 413–421.
- Morrow, G.B.; Whyte, C.S.; Mutch, N.J. Functional Plasminogen Activator Inhibitor 1 Is Retained on the Activated Platelet Membrane Following Platelet Activation. Haematologica 2020, 105, 2824–2833.
- Sillen, M.; Declerck, P.J. A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? Int. J. Mol. Sci. 2021, 22, 2721.
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 364.
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066.
- Wind, T.; Hansen, M.; Jensen, J.K.; Andreasen, P.A. The Molecular Basis for Anti-Proteolytic and Non-Proteolytic Functions of Plasminogen Activator Inhibitor Type-1: Roles of the Reactive Centre Loop, the Shutter Region, the Flexible Joint Region and the Small Serpin Fragment. Biol. Chem. 2002, 383, 21–36.
- Schroeck, F.; Arroyo de Prada, N.; Sperl, S.; Schmitt, M.; Magdolen, V. Interaction of Plasminogen Activator Inhibitor Type-1 (PAI-1) with Vitronectin (Vn): Mapping the Binding Sites on PAI-1 and Vn. Biol. Chem. 2002, 383, 1143–1149.
- Wilczynska, M.; Fa, M.; Ohlsson, P.I.; Ny, T. The Inhibition Mechanism of Serpins. Evidence That the Mobile Reactive Center Loop Is Cleaved in the Native Protease-Inhibitor Complex. J. Biol. Chem. 1995, 270, 29652–29655.
- Lawrence, D.A.; Ginsburg, D.; Day, D.E.; Berkenpas, M.B.; Verhamme, I.M.; Kvassman, J.O.; Shore, J.D. Serpin-Protease Complexes Are Trapped as Stable Acyl-Enzyme Intermediates. J. Biol. Chem. 1995, 270, 25309–25312.
- Boudier, C.; Gils, A.; Declerck, P.J.; Bieth, J.G. The Conversion of Active to Latent Plasminogen Activator Inhibitor-1 Is an Energetically Silent Event. Biophys. J. 2005, 88, 2848–2854.
- Gettins, P.G.W.; Olson, S.T. Inhibitory Serpins. New Insights into Their Folding, Polymerization, Regulation and Clearance. Biochem. J. 2016, 473, 2273–2293.
- Simone, T.M.; Higgins, P.J. Low Molecular Weight Antagonists of Plasminogen Activator Inhibitor-1: Therapeutic Potential in Cardiovascular Disease. Mol. Med. Ther. 2012, 1, 101.
- Dupont, D.M.; Madsen, J.B.; Kristensen, T.; Bodker, J.S.; Blouse, G.E.; Wind, T.; Andreasen, P.A. Biochemical Properties of Plasminogen Activator Inhibitor-1. Front. Biosci. 2009, 14, 1337–1361.
- Fjellström, O.; Deinum, J.; Sjögren, T.; Johansson, C.; Geschwindner, S.; Nerme, V.; Legnehed, A.; McPheat, J.; Olsson, K.; Bodin, C.; et al. Characterization of a Small Molecule Inhibitor of Plasminogen Activator Inhibitor Type 1 That Accelerates the Transition into the Latent Conformation. J. Biol. Chem. 2013, 288, 873–885.
- Lawrence, D.A.; Ginsburg, D.; Olson, S.T.; Palaniappan, S. Engineering Plasminogen Activator Inhibitor 1 Mutants with Increased Functional Stability. Biochemistry 1994, 33, 3643–3648.
- Van De Craen, B.; Declerck, P.J.; Gils, A. The Biochemistry, Physiology and Pathological Roles of PAI-1 and the Requirements for PAI-1 Inhibition in Vivo. Thromb. Res. 2012, 130, 576–585.
- Thorsen, S.; Philips, M.; Selmer, J.; Lecander, I.; Åstedt, B. Kinetics of Inhibition of Tissue-Type and Urokinase-Type Plasminogen Activator by Plasminogen-Activator Inhibitor Type 1 and Type 2. Eur. J. Biochem. 1988, 175, 33–39.
- Lee, E.; Vaughan, D.E.; Parikh, S.H.; Grodzinsky, A.J.; Libby, P.; Lark, M.W.; Lee, R.T. Regulation of Matrix Metalloproteinases and Plasminogen Activator Inhibitor-1 Synthesis by Plasminogen in Cultured Human Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 44–49.
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763.
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090.
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956.
- Oszajca, K.; Bieniasz, M.; Brown, G.; Swiatkowska, M.; Bartkowiak, J.; Szemraj, J. Effect of Oxidative Stress on the Expression of T-PA, u-PA, u-PAR, and PAI-1 in Endothelial Cells. Biochem. Cell Biol. 2008, 86, 477–486.
- Swiatkowska, M.; Szemraj, J.; Al-Nedawi, K.; Pawlowska, Z. Reactive Oxygen Species Upregulate Expression of PAI-1 in Endothelial Cells. Cell. Mol. Biol. Lett. 2002, 7, 1065–1071.
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 Mediates the Expression of Plasminogen Activator Inhibitor-1 via P38 MAPK Pathway in Cultured Human Endothelial Cells. Thromb. Res. 2009, 124, 439–446.
- Orbe, J.; Rodriguez, J.A.; Calvo, A.; Grau, A.; Belzunce, M.S.; Martinez-Caro, D.; Páramo, J.A. Vitamins C and E Attenuate Plasminogen Activator Inhibitor-1 (PAI-1) Expression in a Hypercholesterolemic Porcine Model of Angioplasty. Cardiovasc. Res. 2001, 49, 484–492.
- Gomaa, A.M.S.; Abd El-Mottaleb, N.A.; Aamer, H.A. Antioxidant and Anti-Inflammatory Activities of Alpha Lipoic Acid Protect against Indomethacin-Induced Gastric Ulcer in Rats. Biomed. Pharmacother. 2018, 101, 188–194.
- Martina, V.; Bruno, G.A.; Pannocchia, A.; Zumpano, E.; Tagliabue, M.; Trucco, F.; Giorgianni, A.; Stella, S.; Pescarmona, G.P. PAI-1 Reduction after Treatment with Glutathione in NIDDM. Fibrinolysis 1996, 10, 63–65.
- Bonfigli, A.R.; Pieri, C.; Manfrini, S.; Testa, I.; Sirolla, C.; Ricciotti, R.; Marra, M.; Compagnucci, P.; Testa, R. Vitamin E Intake Reduces Plasminogen Activator Inhibitor Type 1 in T2DM Patients. Diabetes Nutr. Metab. 2001, 14, 71–77.
- Antoniades, C.; Tousoulis, D.; Tentolouris, C.; Toutouza, M.; Marinou, K.; Goumas, G.; Tsioufis, C.; Toutouzas, P.; Stefanadis, C. Effects of Antioxidant Vitamins C and E on Endothelial Function and Thrombosis/Fibrinolysis System in Smokers. Thromb. Haemost. 2003, 89, 990–995.
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH Oxidase in Oxidized LDL-Induced Upregulation of Heat Shock Factor-1 and Plasminogen Activator Inhibitor-1 in Vascular Endothelial Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 104–111.
- Hagiwara, H.; Seki, T.; Ariga, T. The Effect of Pre-Germinated Brown Rice Intake on Blood Glucose and PAI-1 Levels in Streptozotocin-Induced Diabetic Rats. Biosci. Biotechnol. Biochem. 2004, 68, 444–447.
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin Activates the Hypoxia-Inducible Factor-1 Signaling Pathway in Vascular Smooth Muscle Cells. Circ. Res. 2001, 89, 47–54.
- Ren, S.; Shen, G.X. Impact of Antioxidants and HDL on Glycated LDL–Induced Generation of Fibrinolytic Regulators from Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1688–1693.
- Kwon, I.S.; Kim, J.; Rhee, D.K.; Kim, B.O.; Pyo, S. Pneumolysin Induces Cellular Senescence by Increasing ROS Production and Activation of MAPK/NF-ΚB Signal Pathway in Glial Cells. Toxicon 2017, 129, 100–112.
- Vayalil, P.K.; Iles, K.E.; Choi, J.; Yi, A.K.; Postlethwait, E.M.; Liu, R.M. Glutathione Suppresses TGF-Beta-Induced PAI-1 Expression by Inhibiting P38 and JNK MAPK and the Binding of AP-1, SP-1, and Smad to the PAI-1 Promoter. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1281–L1292.
- Dimova, E.Y.; Kietzmann, T. Metabolic, Hormonal and Environmental Regulation of Plasminogen Activator Inhibitor-1 (PAI-1) Expression: Lessons from the Liver. Thromb. Haemost. 2008, 100, 992–1006.
- Libby, P. Inflammation and Cardiovascular Disease Mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S.
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory Processes in Cardiovascular Disease: A Route to Targeted Therapies. Nat. Rev. Cardiol. 2016, 14, 133–144.
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and Cardiovascular Disease: From Mechanisms to Therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130.
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151.
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91.
- Hube, F.; Hauner, H. The Role of TNF-Alpha in Human Adipose Tissue: Prevention of Weight Gain at the Expense of Insulin Resistance? Horm. Metab. Res. 1999, 31, 626–631.
- Takeshita, Y.; Takamura, T.; Hamaguchi, E.; Shimizu, A.; Ota, T.; Sakurai, M.; Kaneko, S. Tumor Necrosis Factor-Alpha-Induced Production of Plasminogen Activator Inhibitor 1 and Its Regulation by Pioglitazone and Cerivastatin in a Nonmalignant Human Hepatocyte Cell Line. Metabolism 2006, 55, 1464–1472.
- Pandey, M.; Loskutoff, D.J.; Samad, F. Molecular Mechanisms of Tumor Necrosis Factor-Alpha-Mediated Plasminogen Activator Inhibitor-1 Expression in Adipocytes. FASEB J. 2005, 19, 1317–1319.
- Macfelda, K.; Weiss, T.W.; Kaun, C.; Breuss, J.M.; Kapeller, B.; Zorn, G.; Oberndorfer, U.; Voegele-Kadletz, M.; Huber-Beckmann, R.; Ullrich, R.; et al. Plasminogen Activator Inhibitor 1 Expression Is Regulated by the Inflammatory Mediators Interleukin-1alpha, Tumor Necrosis Factor-Alpha, Transforming Growth Factor-Beta and Oncostatin M in Human Cardiac Myocytes. J. Mol. Cell. Cardiol. 2002, 34, 1681–1691.
- Hou, B.; Eren, M.; Painter, C.A.; Covington, J.W.; Dixon, J.D.; Schoenhard, J.A.; Vaughan, D.E. Tumor Necrosis Factor Alpha Activates the Human Plasminogen Activator Inhibitor-1 Gene through a Distal Nuclear Factor KappaB Site. J. Biol. Chem. 2004, 279, 18127–18136.
- Samad, F.; Yamamoto, K.; Loskutoff, D.J. Distribution and Regulation of Plasminogen Activator Inhibitor-1 in Murine Adipose Tissue In Vivo. Induction by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. J. Clin. Investig. 1996, 97, 37–46.
- Cigolini, M.; Tonoli, M.; Borgato, L.; Frigotto, L.; Manzato, F.; Zeminian, S.; Cardinale, C.; Camin, M.; Chiaramonte, E.; De Sandre, G.; et al. Expression of Plasminogen Activator Inhibitor-1 in Human Adipose Tissue: A Role for TNF-Alpha? Atherosclerosis 1999, 143, 81–90.
- Papanicolaou, D.A.; Wilder, R.L.; Manolagas, S.C.; Chrousos, G.P. The Pathophysiologic Roles of Interleukin-6 in Human Disease. Ann. Intern. Med. 1998, 128, 127–137.
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356.
- Mestries, J.C.; Kruithof, E.K.O.; Gascon, M.P.; Herodin, F.; Agay, D.; Ythier, A. In Vivo Modulation of Coagulation and Fibrinolysis by Recombinant Glycosylated Human Interleukin-6 in Baboons. Eur. Cytokine Netw. 1994, 5, 275–281.
- Kruithof, E.K.O. Regulation of Plasminogen Activator Inhibitor Type 1 Gene Expression by Inflammatory Mediators and Statins. Thromb. Haemost. 2008, 100, 969–975.
- Toma, I.; McCaffrey, T.A. Transforming Growth Factor-β and Atherosclerosis: Interwoven Atherogenic and Atheroprotective Aspects. Cell Tissue Res. 2012, 347, 155.
- Verrecchia, F.; Mauviel, A. Transforming Growth Factor-β and Fibrosis. World J. Gastroenterol. 2007, 13, 3056.
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β Signalling Drives Vascular Inflammation and Atherosclerosis. Nat. Metab. 2019, 1, 912–926.
- Seeland, U.; Haeuseler, C.; Hinrichs, R.; Rosenkranz, S.; Pfitzner, T.; Scharffetter-Kochanek, K.; Böhm, M. Myocardial Fibrosis in Transforming Growth Factor-Beta(1) (TGF-Beta(1)) Transgenic Mice Is Associated with Inhibition of Interstitial Collagenase. Eur. J. Clin. Investig. 2002, 32, 295–303.
- Grandaliano, G.; Di Paolo, S.; Monno, R.; Stallone, G.; Ranieri, E.; Pontrelli, P.; Gesualdo, L.; Schena, F.P. Protease-Activated Receptor 1 and Plasminogen Activator Inhibitor 1 Expression in Chronic Allograft Nephropathy: The Role of Coagulation and Fibrinolysis in Renal Graft Fibrosis. Transplantation 2001, 72, 1437–1443.
- De Gouville, A.C.; Boullay, V.; Krysa, G.; Pilot, J.; Brusq, J.M.; Loriolle, F.; Gauthier, J.M.; Papworth, S.A.; Laroze, A.; Gellibert, F.; et al. Inhibition of TGF-Beta Signaling by an ALK5 Inhibitor Protects Rats from Dimethylnitrosamine-Induced Liver Fibrosis. Br. J. Pharmacol. 2005, 145, 166–177.
- Kutz, S.M.; Hordines, J.; McKeown-Longo, P.J.; Higgins, P.J. TGF-Beta1-Induced PAI-1 Gene Expression Requires MEK Activity and Cell-to-Substrate Adhesion. J. Cell Sci. 2001, 114, 3905–3914.
- Hirashima, Y.; Kobayashi, H.; Suzuki, M.; Tanaka, Y.; Kanayama, N.; Terao, T. Transforming Growth Factor-Beta1 Produced by Ovarian Cancer Cell Line HRA Stimulates Attachment and Invasion through an up-Regulation of Plasminogen Activator Inhibitor Type-1 in Human Peritoneal Mesothelial Cells. J. Biol. Chem. 2003, 278, 26793–26802.
- Datta, P.K.; Blake, M.C.; Moses, H.L. Regulation of Plasminogen Activator Inhibitor-1 Expression by Transforming Growth Factor-Beta -Induced Physical and Functional Interactions between Smads and Sp1. J. Biol. Chem. 2000, 275, 40014–40019.
- Lund, L.R.; Riccio, A.; Andreasen, P.A.; Nielsen, L.S.; Kristensen, P.; Laiho, M.; Saksela, O.; Blasi, F.; Danø, K. Transforming Growth Factor-Beta Is a Strong and Fast Acting Positive Regulator of the Level of Type-1 Plasminogen Activator Inhibitor MRNA in WI-38 Human Lung Fibroblasts. EMBO J. 1987, 6, 1281–1286.
- Jaffer, O.A.; Carter, A.B.; Sanders, P.N.; Dibbern, M.E.; Winters, C.J.; Murthy, S.; Ryan, A.J.; Rokita, A.G.; Prasad, A.M.; Zabner, J.; et al. Mitochondrial-Targeted Antioxidant Therapy Decreases Transforming Growth Factor-β-Mediated Collagen Production in a Murine Asthma Model. Am. J. Respir. Cell Mol. Biol. 2015, 52, 106–115.
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β Mediates Aortic Smooth Muscle Cell Senescence in Marfan Syndrome. Aging 2019, 11, 3574–3584.
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777.
- García-Trevijano, E.R.; Iraburu, M.J.; Fontana, L.; Domínguez-Rosales, J.A.; Auster, A.; Covarrubias-Pinedo, A.; Rojkind, M. Transforming Growth Factor Beta1 Induces the Expression of Alpha1(I) Procollagen MRNA by a Hydrogen Peroxide-C/EBPbeta-Dependent Mechanism in Rat Hepatic Stellate Cells. Hepatology 1999, 29, 960–970.
- Herrera, B.; Murillo, M.M.; Álvarez-Barrientos, A.; Beltrán, J.; Fernández, M.; Fabregat, I. Source of Early Reactive Oxygen Species in the Apoptosis Induced by Transforming Growth Factor-Beta in Fetal Rat Hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26.
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFbeta1-Induced Suppression of Glutathione Antioxidant Defenses in Hepatocytes: Caspase-Dependent Post-Translational and Caspase-Independent Transcriptional Regulatory Mechanisms. FASEB J. 2003, 17, 1535–1537.
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-Induced Src Kinase and Caveolin-1 Signaling in TGF-Β1-Initiated SMAD2/3 Activation and PAI-1 Expression. PLoS ONE 2011, 6, e22896.
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. P38 MAPK Mediates Fibrogenic Signal through Smad3 Phosphorylation in Rat Myofibroblasts. Hepatology 2003, 38, 879–889.
- Woodward, R.N.; Finn, A.V.; Dichek, D.A. Identification of Intracellular Pathways through Which TGF-Beta1 Upregulates PAI-1 Expression in Endothelial Cells. Atherosclerosis 2006, 186, 92–100.
- Chen, Y.Q.; Su, M.; Walia, R.R.; Hao, Q.; Covington, J.W.; Vaughan, D.E. Sp1 Sites Mediate Activation of the Plasminogen Activator Inhibitor-1 Promoter by Glucose in Vascular Smooth Muscle Cells. J. Biol. Chem. 1998, 273, 8225–8231.
- Marsch, E.; Sluimer, J.C.; Daemen, M.J.A.P. Hypoxia in Atherosclerosis and Inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400.
- Rey, S.; Semenza, G.L. Hypoxia-Inducible Factor-1-Dependent Mechanisms of Vascularization and Vascular Remodelling. Cardiovasc. Res. 2010, 86, 236–242.
- Gao, L.; Chen, Q.; Zhou, X.; Fan, L. The Role of Hypoxia-Inducible Factor 1 in Atherosclerosis. J. Clin. Pathol. 2012, 65, 872–876.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514.
- Lin, M.T.; Kuo, I.H.; Chang, C.C.; Chu, C.Y.; Chen, H.Y.; Lin, B.R.; Sureshbabu, M.; Shih, H.J.; Kuo, M.L. Involvement of Hypoxia-Inducing Factor-1α-Dependent Plasminogen Activator Inhibitor-1 up-Regulation in Cyr61/CCN1-Induced Gastric Cancer Cell Invasion. J. Biol. Chem. 2016, 291, 27433.
- Sanagawa, A.; Iwaki, S.; Asai, M.; Sakakibara, D.; Norimoto, H.; Sobel, B.E.; Fujii, S. Sphingosine 1-phosphate Induced by Hypoxia Increases the Expression of PAI-1 in HepG2 Cells via HIF-1α. Mol. Med. Rep. 2016, 14, 1841–1848.
- Kabei, K.; Tateishi, Y.; Nozaki, M.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Nishide, S.; Uchida, J.; Nakatani, T.; Tomita, S.; et al. Role of Hypoxia-Inducible Factor-1 in the Development of Renal Fibrosis in Mouse Obstructed Kidney: Special References to HIF-1 Dependent Gene Expression of Profibrogenic Molecules. J. Pharmacol. Sci. 2018, 136, 31–38.
- Uchiyama, T.; Kurabayashi, M.; Ohyama, Y.; Utsugi, T.; Akuzawa, N.; Sato, M.; Tomono, S.; Kawazu, S.; Nagai, R. Hypoxia Induces Transcription of the Plasminogen Activator Inhibitor-1 Gene through Genistein-Sensitive Tyrosine Kinase Pathways in Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1155–1161.
- Kimura, D.; Imaizumi, T.; Tamo, W.; Sakai, T.; Ito, K.; Hatanaka, R.; Yoshida, H.; Tsushima, T.; Satoh, K.; Fukuda, I. Hypoxia Enhances the Expression of Plasminogen Activator Inhibitor-1 in Human Lung Cancer Cells, EBC-1. Tohoku J. Exp. Med. 2002, 196, 259–267.
- Toullec, A.; Buard, V.; Rannou, E.; Tarlet, G.; Guipaud, O.; Robine, S.; Iruela-Arispe, M.L.; François, A.; Milliat, F. HIF-1α Deletion in the Endothelium, but Not in the Epithelium, Protects from Radiation-Induced Enteritis. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 15–30.
- Petry, A.; Belaiba, R.S.; Weitnauer, M.; Görlach, A. Inhibition of Endothelial Nitric Oxyde Synthase Increases Capillary Formation via Rac1-Dependent Induction of Hypoxia-Inducible Factor-1α and Plasminogen Activator Inhibitor-1. Thromb. Haemost. 2012, 108, 849–862.
- Görlach, A.; Berchner-Pfannschmidt, U.; Wotzlaw, C.; Cool, R.H.; Fandrey, J.; Acker, H.; Jungermann, K.; Kietzmann, T. Reactive Oxygen Species Modulate HIF-I Mediated PAI-I Expression: Involvement of the GTPase RacI. Thromb. Haemost. 2003, 89, 926–935.
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761.
- Diebold, I.; Djordjevic, T.; Hess, J.; Görlach, A. Rac-1 Promotes Pulmonary Artery Smooth Muscle Cell Proliferation by Upregulation of Plasminogen Activator Inhibitor-1: Role of NFkappaB-Dependent Hypoxia-Inducible Factor-1alpha Transcription. Thromb. Haemost. 2008, 100, 1021–1028.
- An, W.G.; Ahn, Y.-T.; Chua, M.-S.; Whitlock, J.P.; Shin, Y.-C.; Song, W.-H.; Kim, Y.; Eom, C.-Y. Rodent-Specific Hypoxia Response Elements Enhance PAI-1 Expression through HIF-1 or HIF-2 in Mouse Hepatoma Cells. Int. J. Oncol. 2010, 37, 1627–1638.
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular Regulation of the PAI-1 Gene by Hypoxia: Contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J. 2007, 21, 935–949.
- Anfosso, F.; Chomiki, N.; Alessi, M.C.; Vague, P.; Juhan-Vague, I. Plasminogen Activator Inhibitor-1 Synthesis in the Human Hepatoma Cell Line Hep G2 Metformin Inhibits the Stimulating Effect of Insulin. J. Clin. Investig. 1993, 91, 2185–2193.
- Schneider, D.J.; Sobel, B.E. Augmentation of Synthesis of Plasminogen Activator Inhibitor Type 1 by Insulin and Insulin-like Growth Factor Type I: Implications for Vascular Disease in Hyperinsulinemic States. Proc. Natl. Acad. Sci. USA 1991, 88, 9959–9963.
- Schneider, D.J.; Absher, P.M.; Ricci, M.A. Dependence of Augmentation of Arterial Endothelial Cell Expression of Plasminogen Activator Inhibitor Type 1 by Insulin on Soluble Factors Released from Vascular Smooth Muscle Cells. Circulation 1997, 96, 2868–2876.
- Montagnani, M.; Golovchenko, I.; Kim, I.; Koh, G.Y.; Goalstone, M.L.; Mundhekar, A.N.; Johansen, M.; Kucik, D.F.; Quon, M.J.; Draznin, B. Inhibition of Phosphatidylinositol 3-Kinase Enhances Mitogenic Actions of Insulin in Endothelial Cells. J. Biol. Chem. 2002, 277, 1794–1799.
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin Resistance Differentially Affects the PI 3-Kinase- and MAP Kinase-Mediated Signaling in Human Muscle. J. Clin. Investig. 2000, 105, 311–320.
- Alessi, M.C.; Juhan-Vague, I. PAI-1 and the Metabolic Syndrome: Links, Causes, and Consequences. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2200–2207.
- Shimomura, I.; Funahashi, T.; Takahashi, M.; Maeda, K.; Kotani, K.; Nakamura, T.; Yamashita, S.; Miura, M.; Fukuda, Y.; Takemura, K.; et al. Enhanced Expression of PAI-1 in Visceral Fat: Possible Contributor to Vascular Disease in Obesity. Nat. Med. 1996, 2, 800–803.
- Bouarab, C.; Roullot-Lacarrière, V.; Vallée, M.; Le Roux, A.; Guette, C.; Mennesson, M.; Marighetto, A.; Desmedt, A.; Piazza, P.V.; Revest, J.M. PAI-1 Protein Is a Key Molecular Effector in the Transition from Normal to PTSD-like Fear Memory. Mol. Psychiatry 2021, 26, 4968–4981.
- Vaughan, D.E.; Lazos, S.A.; Tong, K. Angiotensin II Regulates the Expression of Plasminogen Activator Inhibitor-1 in Cultured Endothelial Cells. A Potential Link between the Renin-Angiotensin System and Thrombosis. J. Clin. Investig. 1995, 95, 995–1001.
- Rüster, C.; Wolf, G. Angiotensin II as a Morphogenic Cytokine Stimulating Renal Fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199.
- Fogari, R.; Zoppi, A.; Mugellini, A.; Maffioli, P.; Lazzari, P.; Derosa, G. Role of Angiotensin II in Plasma PAI-1 Changes Induced by Imidapril or Candesartan in Hypertensive Patients with Metabolic Syndrome. Hypertens. Res. 2011, 34, 1321–1326.
- Skurk, T.; Lee, Y.M.; Hauner, H. Angiotensin II and Its Metabolites Stimulate PAI-1 Protein Release from Human Adipocytes in Primary Culture. Hypertension 2001, 37, 1336–1340.
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.8K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
01 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No