Obstructive sleep apnea (OSA) is a chronic condition affecting up to one billion people worldwide
[1]. OSA is defined as a sleep-breathing disorder that involves a decrease or complete cessation of airflow despite ongoing efforts to breathe due to a collapsed upper airway. This leads to partial reductions (hypopneas) and complete pauses (apneas) in breathing that usually last between 10 and 30 s, but some may persist longer. This can lead to abrupt reductions in blood oxygen saturation, with oxygen levels falling as much as 40% or more in severe cases
[2]. As a result, several pathological mechanisms ensue such as intermittent hypoxia (IH), sleep fragmentation, episodic hypercapnia, and increased intrathoracic pressure swings
[3][4][5][3,4,5]. Consequently, these processes can induce major changes in the autonomic nervous system balance with both increased tonic and reactive sympathetic activity along with parasympathetic withdrawal, disruption of the hypothalamic–pituitary–adrenal-axis, systemic and cellular oxidative stress, and inflammation, fibrosis, and accelerated cellular senescence, all of which resulted in neurocognitive deficits, endothelial dysfunction, hypertension, and atherosclerosis
[6][7][8][9][10][11][12][13][6,7,8,9,10,11,12,13]. Predictably, OSA is considered as an independent risk factor for cardiovascular disease (CVD) including coronary artery disease (CAD), ischemic stroke, and myocardial infarction (MI)
[14]. The majority of strokes and MIs seem to be prompted by atherothrombotic events along with compromised fibrinolytic activity, increasing the propensity for such events
[15][16][15,16]. The fibrinolytic system is designed to cleave the insoluble polymeric network of fibrin from the vascular system to prevent clot overgrowth and vessel occlusion. Generally, plasminogen is activated by serine proteases plasminogen activators (PAs) including tissue-type PA (tPA) and urokinase-type PA (uPA) into plasmin, which in turn lyses the fibrin and other extracellular matrix components
[17][18][17,18]. To prevent bleeding, plasminogen activator inhibitor-1 (PAI-1) is normally synthesized in equimolar amounts to PAs, forms a covalent bond with Pas, and stabilizes fibrin
[19]. However, many processes including oxidative stress
[20], inflammation
[21], and fibrosis
[22] can lead to elevated levels of PAI-1, which have been implicated in a multitude of diseases and conditions including CVD
[23], cancer
[24], metabolic disease
[25], renal disease
[26], behavioral and psychiatric conditions
[27], and aging processes
[28]. Furthermore, PAI-1 has been shown to induce endothelial dysfunction and atherosclerosis through antifibrinolytic-dependent mechanisms including inflammation
[29], endothelial nitric oxide synthase (eNOS) inhibition
[30], neointimal hyperplasia
[31], and vascular senescence
[28]. Despite the fact that PAI-1 levels are consistently elevated in OSA patients
[32][33][34][35][36][37][38][39][40][41][42][32,33,34,35,36,37,38,39,40,41,42], and that OSA can trigger processes that can upregulate PAI-1 production, little to no attention has been given to PAI-1 as a biomarker or as a promoter of OSA-induced CVD in clinical practice.
2. PAI-1 Sources, Structure, and Function
PAI-1 can be synthetized by numerous types of cells including platelets, macrophages, adipocytes, hepatocytes, vascular smooth muscle cells, endothelial cells, and others
[43][44][45][45,46,47]. Approximately 10% of the PAI-1 produced circulates in the blood or is deposited in the subendothelial matrix, while the rest is retained in platelets
[46][47][48,49]. Platelets can de novo synthesize PAI-1 despite lacking nuclei through activated PAI-1 mRNA, with the synthesis rates being increased upon platelet activation
[48][50]. The circulating PAI-1 fraction exists in its active conformation at levels of 5–50 ng/mL with large intra- and inter-personal variability, while platelet PAI-1 concentrations can reach up to 300 ng/mL with 50% shown to be biologically active
[46][49][50][48,51,52]. Ultimately, PAI-1 plasma levels are increased under numerous pathological conditions
[51][53]. The structure and function of PAI-1 have been extensively reviewed previously
[25][52][53][25,54,55]. Briefly, PAI-1 is a single chain molecule with two interactive domains including a surface-exposed reactive center loop (RCL) that presents as a substrate peptide becoming the primary site for uPA/tPA binding, and a flexible joint region with helices D, E, and F that bind to vitronectin and stabilize PAI-1 in its active form while enhancing its binding affinity to uPA/tPA 200-fold
[24][54][55][56][57][58][59][24,56,57,58,59,60,61]. PAI-1 exists in three distinct structurally and functionally distinct conformations including active, latent, and cleaved
[52][60][54,62]. Unless bound to vitronectin, the active form can be readily converted to the more energetically favorable inactive latent form by internalizing the RCL, which may serve as a regulatory mechanism to prevent excessive anti-fibrinolysis
[61][62][63][63,64,65]. However, the latent form can be reactivated. In its cleaved form, PAI-1 is still able to bind to other proteins with its helix, but its ability to inhibit uPA/tPA is abrogated
[61][63]. As alluded to earlier, PAI-1 is a master regulator of the plasminogen system. PAI-1 can rapidly inactivate uPA/tPA with a second-order-rate constant between 106 and 107 m
−1 s
−1, forming a non-covalent Michaelis-like complex and eventually forming an ester bond between the serine residue of the protease and the carboxyl group of the P1 residue of PAI-1
[64][65][66,67]. PAI-1 also plays an important role in extracellular matrix (ECM) remodeling by indirectly modulating the activity of matrix metalloproteinases (MMPs)
[66][68]. Indeed, by inhibiting the plasmin activation required for the cleavage of pro-MMP, PAI-1 can block ECM degradation
[53][55].
3. Mechanisms Involved in PAI-1 Upregulation
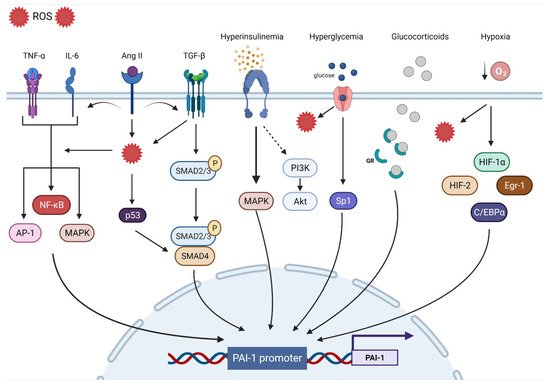
The human PAI-1 promoter shows a high degree of homology with mice and rats, suggesting that they are regulated by similar mechanisms. The 5′-flanking region contains a ‘TATA’ box with several transcription binding sites including hypoxia inducible factor-1α (HIF-1α), Smads, activator protein-1 (AP-1), specificity protein-1 (SP-1), and nuclear factor kappa B (NF-ƘB).
TIn the next section, we will discuss the major contributors to PAI-1 upregulation (
Figure 1).
Figure 1. Transcriptional regulatory pathways implicated in PAI-1 synthesis.
3.1. Oxidative Stress
Oxidative stress (OS) is the end result of an imbalance between the production of oxidants and the capacity of the antioxidant system. Although they play an important role in regulating cellular function and signal transduction, free radicals such as reactive oxygen species (ROS) can be detrimental when produced in excess, given their ability to damage lipids, proteins, and DNA
[67][69]. OS is undeniably a major contributor to multiorgan dysfunction in many disease states including CVD
[68][70]. Indeed, ROS overproduction directly decreases nitric oxide (NO) bioavailability, uncouples eNOS, oxidizes low-density lipoprotein (OxLDL), and induces vascular inflammation
[69][71]. OS is a significant upregulator of PAI-1 transcription. Indeed, incubating endothelial cells with H
2O
2 induced marked increases in PAI-1 mRNA and protein expression
[70][72]. Conversely, the PAI-1 promoter was suppressed by up to 75% in the presence of antioxidants
[71][73]. Furthermore, inhibiting NADPH oxidase, a major source of ROS, abolished the PAI-1 release and promoter activity in cultured endothelial cells
[72][74]. Other experimental in vitro and in vivo studies performed in animal models as well as in humans have shown that the administration of antioxidants can decrease PAI-1 expression
[20][73][74][75][76][77][78][79][80][81][20,75,76,77,78,79,80,81,82,83]. Due to their intricate interactions with multiple signaling pathways and transcription factors, ROS are involved in most of the mechanisms regulating PAI-1 expression. For instance, ROS-induced PAI-1 increased transcription and expression is mediated through the activation of mitogen-activated protein kinase (MAPK) and NF-ƘB pathways that are tightly involved in pro-fibrotic and pro-inflammatory pathways
[72][82][74,84]. ROS signaling can also stimulate AP-1, HIF-1α, and p53, all of which can increase the transcription of PAI-1
[83][84][85,86] (
Figure 1).
3.2. Inflammation
Inflammation is a complex constellation of reactions between the host normal defense processes to internal and external stressors that have been implicated in many conditions and age-related diseases, especially in promoting atherosclerosis, a hallmark of CVD
[85][86][87][87,88,89]. Low-grade inflammation induces endothelial dysfunction and subintimal cholesterol accumulation, leading to the upregulation of intercellular adhesion molecules and selectins that promote the binding and transmigration of inflammatory cells including monocytes and T-helper cells into the vessel wall. Infiltrating monocytes can transform into resident macrophages that express and activate inflammasomes that are key to the propagation of inflammation through the generation of multiple cytokines that amplify the inflammatory cascade within the vessel wall
[87][89]. Coupled with enhanced ROS production, inflammation enters a vicious cycle in combination with OS, further aggravating atherosclerosis
[88][90]. The link between inflammation and the fibrinolytic system is well-established. Experimental in vitro and in vivo studies as well as clinical studies have identified tumor necrosis factor-α (TNF-α) as a substantial contributor to increased PAI-1 expression
[89][90][91][92][93][94][91,92,93,94,95,96]. In endothelial cells, TNF-α upregulated PAI-1 levels and was abolished by N-acetyl cysteine, indicating ROS as a mediator
[71][73]. Administration of TNF-α in mice significantly increased the PAI-1 levels in adipose tissue, while obese mice treated with antibodies targeting TNF-α exhibited reduced plasma PAI-1 expression and adipose tissue-PAI-1 levels
[95][96][97,98]. It is suggested that TNF-α can induce PAI-1 gene expression via redox-sensitive mechanisms triggering NF-ƘB translocation and interaction with a regulatory region that is present on the PAI-1 promoter
[21][94][21,96]. These data showcase the interplay between inflammation and OS and their integral role in upregulating PAi-1. Other pathways have been suggested in TNF-α-mediated PAI-1 induction including MAPK and protein kinase C
[91][93]. Interleukin-6 (IL-6) is another inflammatory cytokine involved in PAI-1 upregulation. IL-6 is an acute phase inflammatory reaction protein that can induce C-reactive protein (CRP) synthesis and cortisol production
[97][99]. Animals injected with IL-6 had significant increases in PAI-1 levels, while using IL-6 receptor antagonist reduced the PAI-1 expression in COVID patients
[98][99][100,101]. IL-6 can activate NF-ƘB and MAPK, leading to increased PAI-1 transcription
[53][100][55,102] (
Figure 1).
3.3. Fibrosis
Progressive vascular fibrosis is a prominent feature of atherosclerosis and CVD
[101][103]. Transforming growth factor-β (TGF-β) is a major regulator of the fibroproliferative response to tissue damage
[102][104]. TGF-β can control cell proliferation and migration, matrix synthesis, calcification, and immunomodulation, all being integral components of atherosclerosis
[103][105]. TGF-β can be produced by all cells composing the vasculature and can also be produced in atherosclerotic lesions. However, TGF-β is mainly released by activated platelets adherent to activated endothelium. As a result, TGF-β induces the transcription of platelet-derived growth factor, collagens, fibronectin, and thrombospondins while suppressing the breakdown of ECM by inducing the transcription of PAI-1 and metalloprotease inhibitors, leading to the accumulation of the fibrotic matrix followed by calcification
[101][103][103,105]. Overall, TGF-β production in atherosclerotic lesions can result in negative remodeling and progressive narrowing of the arteries, leading to MI and stroke
[101][103]. TGF-β is considered as one of the major drivers of PAI-1 upregulation. In vitro studies have shown that PAI-1 expression is induced by TGF-β in various types of cells, while elevated PAI-1 levels are associated with enhanced TGF-β expression and ECM deposition under many pathological conditions
[22][104][105][106][107][108][109][22,106,107,108,109,110,111]. TGF-β can induce PAI-1 production through the activation of the Smad pathway via the nuclear translocation of the Smad 2/3 and Smad 4 complex and binding to the PAI-1 promoter
[110][112]. Interestingly, TGF-β can induce ROS production and suppress antioxidant activity in various types of cells and in vivo
[111][112][113][114][115][116][117][113,114,115,116,117,118,119]. Thus, PAI-1 expression can also be mediated through TGF-β-induced ROS production. MAPK and NF-ƘB signaling are redox sensitive pathways that can be induced by TGF-β
[53][112][118][119][55,114,120,121]. In TGF-β treated cells, inhibition of NADPH oxidase blocked TGF-β induced MAPK activated PAI-1 expression
[83][85]. Furthermore, TGF-β can upregulate PAI-1 through Smad interactions with p53 and the transcription factors AP-1 and SP-1
[22][83][120][22,85,122] (
Figure 1).
3.4. Hypoxia
Hypoxia triggers many cellular processes both in physiological and pathological conditions and has been associated with vascular dysfunction and atherosclerosis
[121][123]. Vascular wall cells respond to hypoxia by tuning metabolism, angiogenesis, inflammation, cell survival signaling, and ultimately, may develop endothelial dysfunction
[122][123][124,125]. The main regulator of such processes is the transcription factor HIF-1α. Under normoxic conditions, HIF-1α is constantly degraded, whereas hypoxia promotes its stability and transcriptional activity
[124][126]. However, HIF-1α is stabilized in atherosclerotic lesions even under normoxic conditions. ROS, OxLDL, NF-ƘB, and other factors are promoted by HIF-1α and in return, enhance HIF-1α stability
[121][123]. PAI-1 is one of the main transcriptional targets of HIF-1α. Indeed, cells exposed to hypoxia display increased PAI-1 mRNA expression and stability
[125][126][127][128][129][127,128,129,130,131]. HIF-1α knockdown limited irradiation-induced PAI-1 upregulation in endothelial cells
[130][132]. ROS production in endothelial cells induced HIF-1α and subsequently PAI-1 production
[131][132][133,134]. Additionally, ROS induced HIF-1α via a specific NF-ƘB binding site in the HIF-1 promoter
[133][135]. Indeed, upregulation of the pulmonary artery smooth muscle PAI-1 was induced by an NF-ƘB-dependent HIF-1α transcription
[134][136]. Although HIF-1α appears to dominate the PAI-1 transcriptional response to hypoxia, other pathways including HIF-2α, early growth response protein-1 (Egr-1), and CCAAT-enhancer-binding protein-α (C/EBPα) can augment this response independently of HIF-1α
[135][136][137,138] (
Figure 1).
3.5. Hormones
Insulin can directly stimulate PAI-1 production in hepatocytes, an effect that is augmented by the presence of insulin-like growth factor
[137][138][139,140]. The same effect was observed in cocultured endothelial cells and smooth muscle cells (SMCs)
[139][141]. In the context of insulin resistance, compensatory hyperinsulinemia decreases the activity of the PI3-K/Akt pathway and augments the MAPK/ERK pathway, a major driver of PAI-1 production
[140][141][142,143]. Elevated levels of glucose can also directly increase the expression of PAI-1 in endothelial cells and SMC through an effect on two adjacent Sp1 sites
[120][122]. These data explain the elevated levels of PAI-1 in conditions characterized by hyperinsulinemia and hyperglycemia such as obesity, metabolic syndrome, and type 2 diabetes mellitus
[25][142][143][25,144,145]. Under intense stress, very high levels of glucocorticoid hormones can increase the production of PAI-1 protein
[144][146]. Glucocorticoids bind to their cytoplasmic glucocorticoid receptor and the complex is translocated to the nucleus and directly binds to the glucocorticoid response element that enhances PAI-1 transcription
[84][86]. Angiotensin II, a major vasoconstrictor and contributor to hypertension upregulated by the activation of the renin–angiotensin–aldosterone system (RAAS), has been reported to induce PAI-1 expression in cultured endothelial cells in an angiotensin receptor independent manner
[145][147]. Ang II can increase ROS production, fibrotic signaling (TGF-β), and inflammation, all of which can increase the expression of PAI-1
[146][147][148][148,149,150] (
Figure 1).