 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ALBERTO FALCHETTI | + 3776 word(s) | 3776 | 2021-01-14 06:54:49 | | | |
| 2 | Dean Liu | -1018 word(s) | 2758 | 2021-01-28 09:17:05 | | | | |
| 3 | Dean Liu | Meta information modification | 2758 | 2021-02-02 04:42:58 | | |
Video Upload Options
It is well consolidated that a common mesenchymal cell progenitor shared by bone, skeletal muscle, and adipocytes cell progenitors exists. This makes the role of the skeleton in energy metabolism no longer surprising. Moreover, it also suggests that bone fragility could also be seen as a consequence of a “poor” quality in nutrition. Despite of the fact that ketogenic diet has been proven to be effective in epilepsy, long-term follow-up studies on epileptic children undergoing ketogenic diet reported an increased incidence of bone fractures and decreased bone mineral density too. However, the causes of such negative impacts on bone health have to be better defined. More recently, shorter-terms, or repeated cyclically for more or less short periods, KDs are used also in patients affected by T2DM, obesity, PCOS and so on. While possible correlations between KD and bone health were at least conceivable in children, it remains unclear whether the KD effects on bone health may be different in adults. Only few clinical studies have been adequately designed to investigate bone health are scarce and bone health related aspects, so far. A narrative review on this issue, together practical advice on designing and implementing clinical studies on ketogenic nutritional regimens and bone health outcomes, is provided.
1. Introduction
During the evolution, the mankind has gone through alternating periods of famine/abundances, determined by the seasons and environmental condition changes, consequently inducing switching metabolism efficiency. Of course, the capacity of adaptation and adjustment to these changes has helped us to survive as a species. Currently, in developed countries, radical diet fluctuations are extremely rare, and in this sense, the human metabolism is largely “unchallenged”, but whether or not this represents a favorable aspect is hard to assess. To date, an important aspect to be considered is not only the quantity of ingested food, but also its quality and nutrients combination. Indeed, obesity and type 2 diabetes mellitus (T2DM) are approaching epidemic proportions, which is also a consequence of a “poor” quality in nutrition. Through the metabolism, our body draws the necessary energy from food, during both rest and physical activity. The nutrients in food, summarized for convenience in carbohydrates (CHO), proteins and fats, are broken down during digestion into smaller molecules: glucose, amino acids, and fatty acids, respectively. CHO are the main source of energy for the human body. Interconnections between glycolysis, tricarboxylic acids cycle (TCA) and ketogenesis exist, as summarized in Figure 1. Each oxidized glucose molecule degraded within the glycolysis pathway may provide an effective yield of 30–32 molecules of the most useful energy molecule known as adenosine triphosphate (ATP). Additionally, proteins and lipids degradation contribute to ATP formation. The term “ketogenic”, in general, indicates the capacity to stimulate the production of ketone bodies (KBs) by various cells in our body, when a supplementation of less than 100 g of glucose is provided in the diet.The most recent knowledge of bone pathophysiology shows evidence of the role of the skeleton in energy metabolism as well, in particular in glucose homeostasis. However, both basic and clinical studies on the possible relationship between ketogenic nutritional regimens and skeletal health are still needed, which is also in consideration of the vast “audience” of patients for whom ketogenic diets (KDs) can be of great help, other than epileptic subjects resistant to specific therapies.
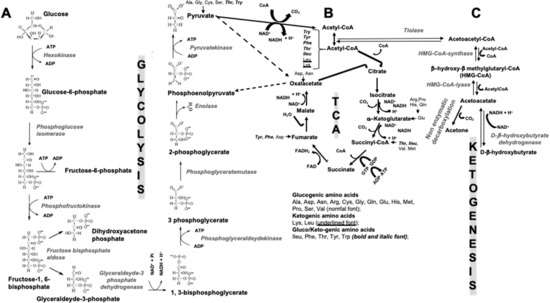
Figure 1. Scheme of interconnections between glycolysis (A), tricarboxylic acids cycle (TCA) (B), and ketogenesis (C). ADP = adenosine diphosphate; ATP = adenosine triphosphate; Acetyl-CoA = acetyl coenzyme A; CO2 = carbon dioxide; CoA = coenzyme A; FAD = flavin adenine dinucleotide; GDP = Guanosine diphosphate; GTP = Guanosine triphosphate; H+ = positively charged hydrogen ion, i.e., a hydrogen atom deprived of its electron; HMG-CoA = 3-hydroxy-3-methyl-glutaryl-coenzyme A; NAD = nicotinamide adenine dinucleotide; NADH = reduced version of NAD; Pi = inorganic phosphate; at the bottom of Figure 1B (under the TCA) classification of glucogenic-, ketogenic-, and gluco/ketogenic amino acids is reported together with the specification of the font type in which they are written.
2. Ketogenic Diets
KDs are normal-high-fat and low-CHO diets that respect an adequate protein regimen, and this proved to be effective in epilepsy in the last 100 years, with potential mechanisms including the direct anticonvulsant effect and the reduced neuronal excitability induced by KBs. Interestingly, it has been shown that KBs produce more energy than glucose due to the metabolic effects of ketosis. In particular, the high chemical potential of 3-β-hydroxybutyrate (3βOHB), the major KB in human metabolism, leads to an increase in the Gibbs free energy (∆G0), or the thermodynamic potential that is minimized when a system reaches chemical equilibrium at constant pressure and temperature[1], from the hydrolysis of ATP molecules. The blood sugar levels, although reduced, remain within the physiological range since glucose in blood comes from two sources: (a) the glucogenic amino acids [alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, histidine, methionine, proline, serine, and valine (Figure 1)], and (b) the glycerol released from triglycerides of lysine residue (lysine is a ketogenic amino acid (Figure 1)).
In long-term follow-up studies, approximately 20% of children treated with KD (≥6 years) experienced an increased incidence of bone fractures[2]. Bone fractures and decreased bone mineral density (BMD) are of concern for children maintained on KD therapy (KDT) and chronic antiepileptic drugs (AED)[2][3][4][5][6]. The causes of such negative effects on bone health may be due to the KD itself, to the high “acid load” milieu via the KBs, to alterations in vitamin D levels, to decreased levels of growth factors, together with both the concomitant use of AEDs, and the reduced/absent mobilization observed in a large proportion of patients with intractable epilepsies[5][6].
2.1. Forms of KDs
Schematically, four major forms of KDs can be described: (1) the “classic” KD; (2) the modified Atkins diet (MAD); (3) the medium chain triglyceride diet (MCT); and (4) the low glycemic index diet (LGID). These regimens are mainly differentiating in the lipids to the protein/CHO ratio[7]. More recently, Very Low Calories Ketogenic Diet (VLCKD) has been developed as a nutritional intervention mimicking fasting through a marked restriction of daily intake of CHO, usually <30 g/day (≃13% of total energy intake), with a relative increase in the proportions of fats (≃44%) and proteins (≃43%) and a total daily energy intake <800 Kcal. Importantly, VLCKD should not be considered as a hyper-protein diet since its daily protein intake is around 1.2–1.5 g/kg of ideal body weight[8]. Weight loss in obese subjects is able to reduce the prevalence of related complications, in terms of type 2 diabetes mellitus (T2DM) prevention and severity, hypertension, dyslipidemia, sleep apnea, fatty liver disease, osteoarthritis, stress incontinence, gastroesophageal reflux, and polycystic ovary syndrome[8]. Consequently, several guidelines, including the Italian ones, have listed specific metabolic diseases in which VLCKD is indicated in the clinical management of patients. Unfortunately, due to the lack of adequate studies, bone and mineral metabolism diseases are still not considered as possibly influenced by KD[8].
2.2. Are KBs the Only Main Source of Energy in Fast and/or Low Carbohydrate Periods?
During a prolonged fasting, gluconeogenesis removes intermediate products from the Krebs cycle or TCA (Figure 1) and directs acetyl-coenzyme A (CoA) towards KBs, which may also have a role in regulating food intake. The main KBs in humans are represented by 3βOHB, acetone and acetoacetic acid (Figure 1) generated by the liver from fatty acids[9] during periods of low food intake (fasting), carbohydrate restrictive diets, starvation, and prolonged intense physical exercise[10]. They represent ancient fuel substrates, evolutionarily preserved, and a fundamental energy source for heart, skeletal muscle, kidney, and brain. It has been suggested that KBs, in particular 3βOHB, preserve muscle protein by the occurrence of systemic inflammation, also participating to the metabolic defense against insulin-induced hypoglycemia[11]. Moreover, 3βOHD is not only the substrate for ATP production (Figure 2), but binding to specific hydroxyl-carboxylic acid receptors (HCAR), inhibits histone deacetylase (HDAC) enzymes, free fatty acid receptors (FFAR), and the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, thus acting as a signaling molecule, promoting the transcription of genes for oxidative stress resistance factors[12]and ultimately influencing, at an epigenetic level, bone cell physiology[11] (Figure 3).
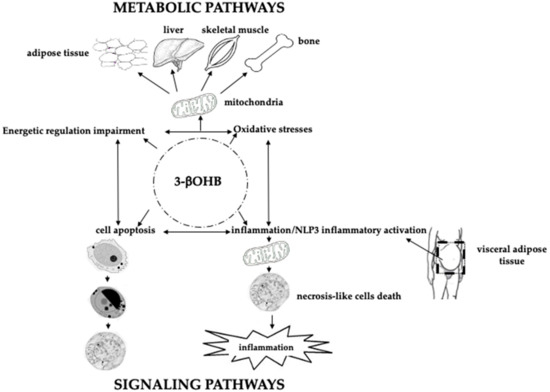
Figure 2. Schematic role of 3betahydroxybutyrate (3βOHB) in energy metabolism, response to oxidative stress, apoptosis, inflammation and signaling pathways. In the center of the figure, within the dotted circle, is depicted the molecule of 3βOHB, “central” to all the physiological events described. The top of the figure shows how 3βOHB, “through the intervention of mitochondria”, may impact the metabolism of adipose tissue, liver, skeletal muscle, and bone, both in terms of oxidative stress modulation and energetic regulation impairment. In turn, oxidative stress, and energetic regulation impairment impact, even with 3βOHB, on cell apoptosis, and inflammation/NLP3 inflammatory activation, (lower figure). Visceral adipose tissue plays an active role on the above events. Thus, 3βOHB may act as a signaling molecule.
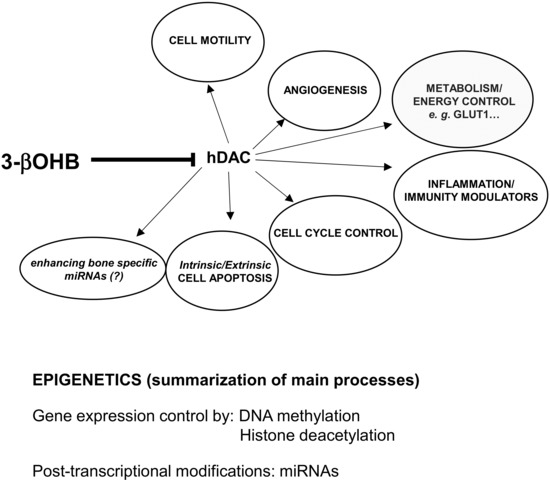
Figure 3. Molecular links between biological functions, environmental factors and 3betahydroxybutyrate (3βOHB). 3βOHD inhibits the function of histone deacetylases (hDAC), molecules representing key transcriptional cofactors that regulating gene expression by deacetylation of lysine residues on histone and nonhistone proteins. Activity of hDAC is important to regulate/influence all the physiopathological events inserted inside the ellipsoidal figures. Through the influence exerted on the processes of DNA methylation, histonic deacetylation and microRNAs expression, 3βOHB may act at epigenetic levels, mostly involving changes that affect gene activity and expression.
3. Skeleton and Energy Metabolism: A Complex “Multiplayers Ping Pong Match Model”
Bone cell progenitors, skeletal muscle cells and adipocytes share a common mesenchymal stem cell progenitor, and the bone cell progenitors can be redirected to become fat cells. These mechanisms evolved to acquire and store fuel, and so it is no longer surprising that the skeleton could also play a role in energy metabolism. In the early tetrapods, a vertebrate superclass with four limbs prevalently adapted to life in a subaerial environment; the evolution of a large appendicular skeleton, powered by robust skeletal muscles, was a successful strategy for walking on the ground, and this general concept can also be extended to mankind. This strategy also justified the need for a “new” skeleton as storage for the calcium contemporary, providing hormonal/molecular mechanisms for the “fast” calcium-phosphate removal from the bone. The development of the parathyroid glands, producing the parathyroid hormone (PTH), was equally necessary. According to its needs, our body may orchestrate different appropriate energy strategies to determine or imbalance with the loss of bone mass (and osteoporosis) or muscle/fat disorders[13]. Over the past 25 years, some findings have suggested human skeleton to play a role in energy metabolism through “local” hormones, such as adipokines, Insulin/Insulin-like growth factor-1 (IGF-1), osteocalcin (OC)/undercarboxylated osteocalcin (UcOC) pathways[13][14][15], and bone morphogenic proteins (BMPs)[16], in cooperation with organs involved in metabolic control (e.g., skeletal muscles, small intestine, and endocrine pancreas)[13]. This complex molecular multidirectional network may be fundamental in maintaining energy homeostasis by controlling and coordinating both “fuel” uptake and energy expenditure, probably within an equally complex hierarchical order in which the central nervous system and peripheral energy centers, sensing and regulating the energy needs, cooperate.
3.1. Bone Remodeling Needs of Boosting Energy
Bone remodeling is a continuous skeletal metabolic structural adaptation to stress forces, fundamental to always ensure a “dynamic” bone structure to adequately satisfy the different biomechanical needs throughout life. To be effective, bone remodeling must have an extremely coordinated regulation, in time and space, of bone resorption and new bone formation phases[17][18], by systemic and local release of cytokines and growth factors[13][14][15][16]. Osteoblasts (OBLs), lining cells, osteoclasts (OCLs), and osteocytes (Ocs) are ultra-organized into the so called “bone remodeling units” (BRUs) with a highly regulated coupling of the specific bone cell functions[19]. There are approximately two million BRUs working in each individual at any given time. The first step of the remodeling cycle consists of bone resorption by OCLs, the “pick hammers”, which release protons and specific proteases (an energy consuming process), thus creating a highly acid environment, which is an essential prerequisite for bone resorption. Later, stromal-derived mesenchymal cells, recruited at the newly excavated site, differentiate into OBLs, the “bricklayers”, and are able to affix and mineralize the “new” freshly formed bone[20] by the production and secretion of specific proteins, particularly type I collagen. All these biological processes require adequate and appropriate energy expenditure. Once bone formation is completed, OBLs give rise to Ocs[21] that act as mechanical sensors [22] and secrete specific osteokines, such as sclerostin. Sclerostin binds to the low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) receptors on OBLs cell surface and inhibits the Wnt signaling pathway determining anti-anabolic effects on bone formation[23]. In humans, the complete remodeling process takes approximately 100 days and the entire skeleton is remodeled every 10 years. Consequently, a high energy cost must be incurred[24].
3.2. Insulin, Osteocalcin, Osteoprotegerin, Receptor Activator of Nuclear Factor Kappa-B Ligand: A Complex, Interconnected, Network
At the skeletal level, insulin signaling stimulates either the OC expression or the OBLs differentiation, by inhibiting Twist2, an inhibitor of the osteoblastogenic factor Runt-related transcription factor 2 (RUNX2), also known as the core-binding factor subunit alpha-1 (CBF-alpha-1), encoded by the RUNX2 gene[25]. The expression of Forkhead box protein O1 (Foxo1), a transcription factor (TF) encoded by FOXO1 gene[26], is higher in skeletal tissues and both Foxo1 activity and the expression increases in mouse mesenchymal cells under osteogenic stimulants. The Foxo1 silencing blocks the expression of Runx2 as well as other osteogenic markers, such as alkaline phosphatase (ALP) and OC, thus reducing the calcification process, even in the presence of strong osteogenic stimulants. Therefore, the main mechanism, through which Foxo1 affects mesenchymal cell differentiation into OBLs, occurs exactly through the regulation of Runx2[27]. Moreover, Foxo1 plays important roles in the regulation of both gluconeogenesis and glycogenolysis by insulin signaling and it is also central to the decision for preadipocytes to commit to adipogenesis [28]. This TF is primarily regulated through phosphorylation on multiple residues, and its transcriptional activity is dependent on its phosphorylation state, resulting in the accumulation of carboxylate OC in the bone matrix. Conversely, insulin activates OCLs and accelerate bone turnover via the increasing of the ratio of the osteoprotegerin/receptor activator of nuclear factor kappa-B ligand (OPG/RANKL). Activation of OCLs determines decarboxylation of the bone matrix-embedded OC, which, in turn, reduce OC affinity to the matrix itself with consequent UcOC release into the circulation. The circulating UcOC stimulates the expression of insulin in pancreas and adiponectin in adipose tissue, both expressing the cytosolic OC specific receptor, GPRC6A, also expressed in testis tissue[29]. Table 1 describes the main hypothesized endocrine UcOC functions.
Table 1. Endocrine effects/functions of UcOC, mediated by binding to specific GPRC6A membrane receptor[15], on insulin, energy, and testosterone metabolism.
| Effects on Insulin Metabolism | Effects on Energy Metabolism and Other Suggested Endocrine Effects |
|---|---|
| Increase of β-cells proliferation, synthesis, and secretion of insulin | Improvement of energy expenditure through multiple mechanisms[15]. |
| Improvement of insulin sensitivity through multiple mechanisms[15]. | Increased expression of adiponectin (ADPN) in white fat and reducing lipid accumulation (and inflammation in the steatosis liver). |
| Modulation of hepatic insulin sensitivity | Stimulation of energy expenditure by increasing mitochondrial biogenesis in muscle and regulating the expression of genes involved in energy consumption in brown adipose and musculoskeletal tissue. |
| Promotion of male fertility by stimulating the synthesis of testosterone in Leydig cells. |
Interestingly, genetic data support the fact that, through UcOC, the skeleton contributes to regulating glucose metabolism: (i) heterozygous dominant negative mutation of GPRC6A gene in two patients has been associated with peripheral testicular failure, glucose intolerance, insulin resistance, and increased body mass index (BMI)[30]; (ii) a significant association between BGLAP (OC gene) genetic variants and BMI in healthy subjects has been reported to be most likely associated with body mass as composite phenotype and less likely associated with adipose tissue itself, even though not only the BGLAP gene variants may cause this association[31]; (iii) R94Q Single Nucleotide Polymorphism (SNP) of BGLAP exon 4, near to one γ-carboxylation site of OC, associates with insulin sensitivity and glucose disposal in Afro-Americans[32]. However, DNA polymorphic variants of BGLAP have been excluded as a major risk factor for T2DM in Caucasians[24]; and (iv) patients with autosomal dominant osteopetrosis, due to an OCL activity deficit, exhibited reduced levels of UcOC together with hypoinsulinemia[33]. Interestingly, if exposed to chronic high glucose levels, the bone marrow stromal stem cells (BMSCs) show an enhanced adipogenic activity due to both the enhancement of the peroxisome proliferator-activated receptor gamma (PPARγ)-dependent pathway, and the of cyclin D3 expression increase[34], which, in turn, is associated with a decreased Runx2[35], ALP[36], and OC expression in OBLs.
In summary, the skeleton can achieve its role in maintaining glucose homeostasis through the interaction with different systems/organs, such as endocrine glucose-regulation of the pancreas, liver, white adipose and skeletal muscle tissue, especially when a general increase in fuel requirements is necessary.
References
- Perrot, P. A to Z of Thermodynamics; Oxford University Press: Oxford, UK, 1998.
- Groesbeck, D.K.; Bluml, R.M.; Kossoff, E.H. Long-term use of the ketogenic diet in the treatment of epilepsy. Dev. Med. Child Neurol. 2007, 48, 978–981.
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and Late-onset Complications of the Ketogenic Diet for Intractable Epilepsy. Epilepsia 2004, 45, 1116–1123.
- Bergqvist, A.C.; Schall, J.I.; Stallings, V.A. Vitamin D Status in Children with Intractable Epilepsy, and Impact of the Ketogenic Diet. Epilepsia 2007, 48, 66–71.
- Bergqvist, A.C.; Schall, J.I.; Stallings, V.A.; Zemel, B.S. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am. J. Clin. Nutr. 2008, 88, 1678–1684.
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Bergqvist, A.C.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192.
- Caprio, M.; the Cardiovascular Endocrinology Club of the Italian Society of Endocrinology; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386.
- Daniel, S.; Soleymani, T.; Garvey, W.T. A complications-based clinical staging of obesity to guide treatment modality and intensity. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 377–388.
- Stryer, L. Biochemistry, 4th ed.; W.H. Freeman and Company: New York, NY, USA, 1995; pp. 510–515, 581–613, 775–778.
- Koeslag, J.H.; Noakes, T.D.; Sloan, A.W. Post-exercise ketosis. J. Physiol. 1980, 301, 79–90.
- Møller, N. Ketone Body, 3-Hydroxybutyrate: Minor Metabolite—Major Medical Manifestations. J. Clin. Endocrinol. Metab. 2020, 105, 2884–2892.
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by -Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214.
- Devlin, M.J.; Rosen, C.J. The bone–fat interface: Basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol. 2015, 3, 141–147.
- Kanazawa, I. Osteocalcin as a hormone regulating glucose metabolism. World J. Diabetes 2015, 6, 1345–1354.
- Ferron, M.; Lacombe, J. Regulation of energy metabolism by the skeleton: Osteocalcin and beyond. Arch. Biochem. Biophys. 2014, 561, 137–146.
- Sánchez-Duffhues, G.; Hiepen, C.; Knaus, P.; Dijke, P.T. Bone morphogenetic protein signaling in bone homeostasis. Bone 2015, 80, 43–59.
- Nakahama, K.-I. Cellular communications in bone homeostasis and repair. Cell. Mol. Life Sci. 2010, 67, 4001–4009.
- Tsukamoto, I.; Hie, M.; Iitsuka, N.; Otsuka, T. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int. J. Mol. Med. 2011, 28, 455–462.
- Zaidi, M. Skeletal remodeling in health and disease. Nat. Med. 2007, 13, 791–801.
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1–induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765.
- Rey, C.; Combes, C.; Drouet, C.; Glimcher, M.J. Bone mineral: Update on chemical composition and structure. Osteoporos. Int. 2009, 20, 1013–1021.
- Bonewald, L.F.; Kneissel, M.; Johnson, M. Preface: The Osteocyte. Bone 2013, 54, 181.
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J. Biol. Chem. 2005, 280, 19883–19887.
- Lee, W.-C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266.
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235.
- Lucero, C.M.; Vega, O.A.; Osorio, M.M.; Tapia, J.C.; Antonelli, M.; Stein, G.S.; Van Wijnen, A.J.; Galindo, M. The cancer-related transcription factor Runx2 modulates cell proliferation in human osteosarcoma cell lines. J. Cell. Physiol. 2013, 228, 714–723.
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a Novel Regulator of Osteoblast Differentiation and Skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065.
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs, W.H.; Arden, K.C.; Accili, D. The Forkhead Transcription Factor Foxo1 Regulates Adipocyte Differentiation. Dev. Cell 2003, 4, 119–129.
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in β-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683.
- Oury, F.; Ferron, M.; Huizhen, W.; Confavreux, C.; Xu, L.; Lacombe, J.; Srinivas, P.; Chamouni, A.; Lugani, F.; Lejeune, H.; et al. Osteocalcin regulates murine and human fertility through a pancreas-bone-testis axis. J. Clin. Investig. 2013, 123, 2421–2433.
- Korostishevsky, M.; Malkin, I.; Trofimov, S.; Pei, Y.; Deng, H.-W.; Livshits, G. Significant association between body composition phenotypes and the osteocalcin genomic region in normative human population. Bone 2012, 51, 688–694.
- Das, S.K.; Sharma, N.K.; Elbein, S.C. Analysis of Osteocalcin as a Candidate Gene for Type 2 Diabetes (T2D) and Intermediate Traits in Caucasians and African Americans. Dis. Markers 2010, 28, 281–286.
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; Depinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell 2010, 142, 296–308.
- Wang, A.; Midura, R.J.; Vasanji, A.; Wang, A.J.; Hascall, V.C. Hyperglycemia Diverts Dividing Osteoblastic Precursor Cells to an Adipogenic Pathway and Induces Synthesis of a Hyaluronan Matrix That Is Adhesive for Monocytes. J. Biol. Chem. 2014, 289, 11410–11420.
- García-Hernández, A.; Arzate, H.; Gil-Chavarría, I.; Rojo, R.; Moreno-Fierros, L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone 2012, 50, 276–288.
- Ehnert, S.; Thomas, F.; Ihle, C.; Mayer, L.; Braun, B.; Graeser, J.; Flesch, I.; Stöckle, U.; Nussler, A.K.; Pscherer, S. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation —Description of a novel in vitro model. Exp. Cell Res. 2015, 332, 247–258.


